Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States.
Department of Biochemistry and Molecular Genetics, University of Louisville School of Medicine, Louisville, KY, United States.
Front Immunol. 2020 Sep 23;11:2136. doi: 10.3389/fimmu.2020.02136. eCollection 2020.
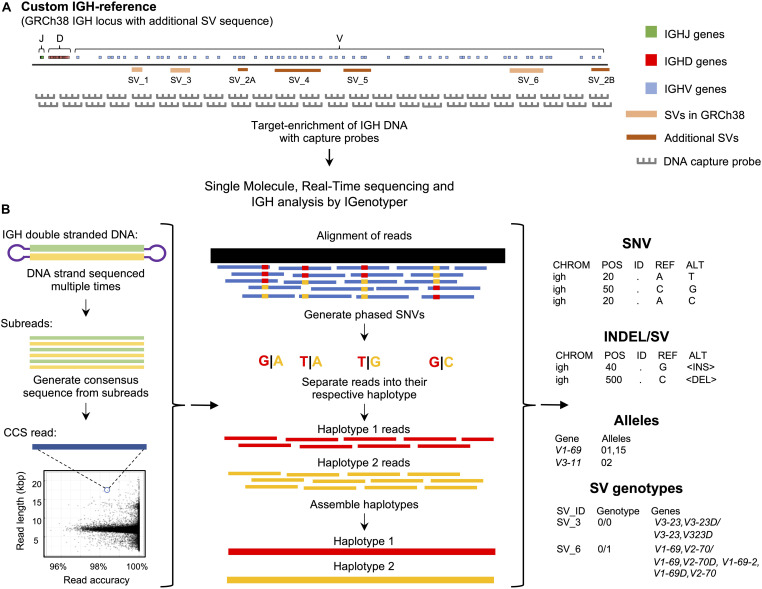
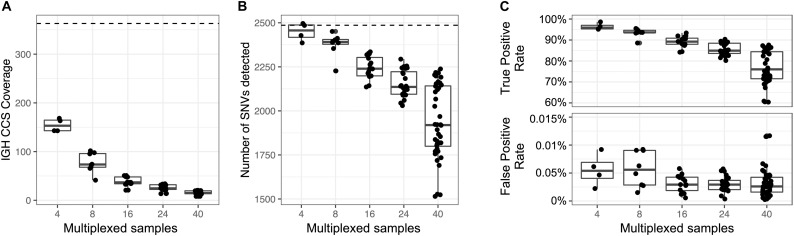
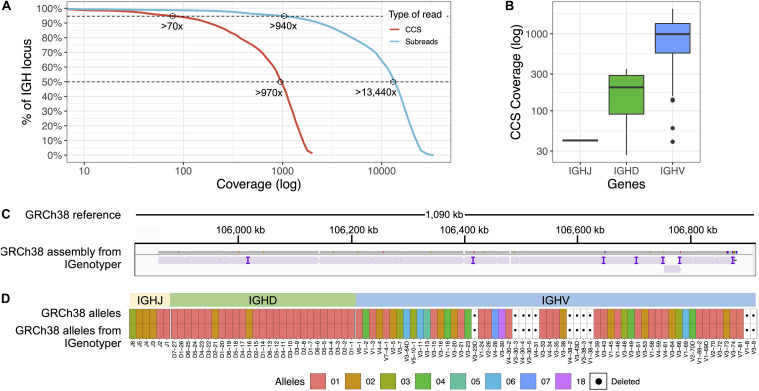
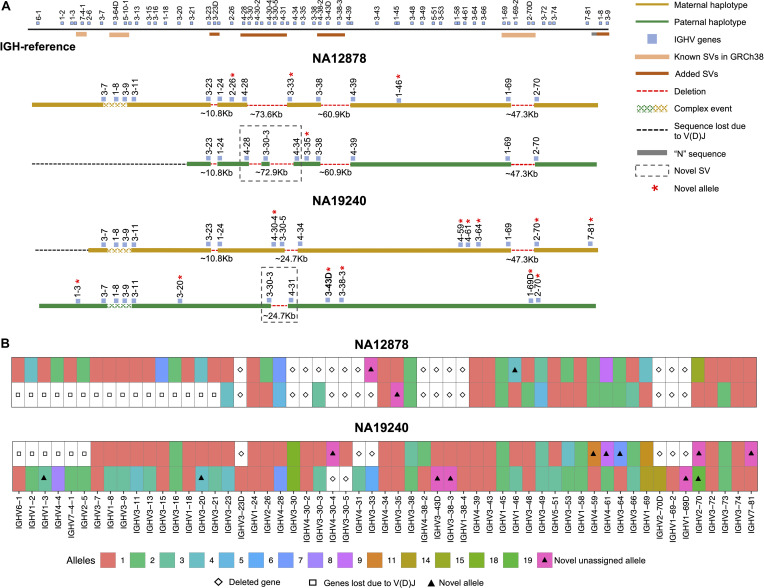
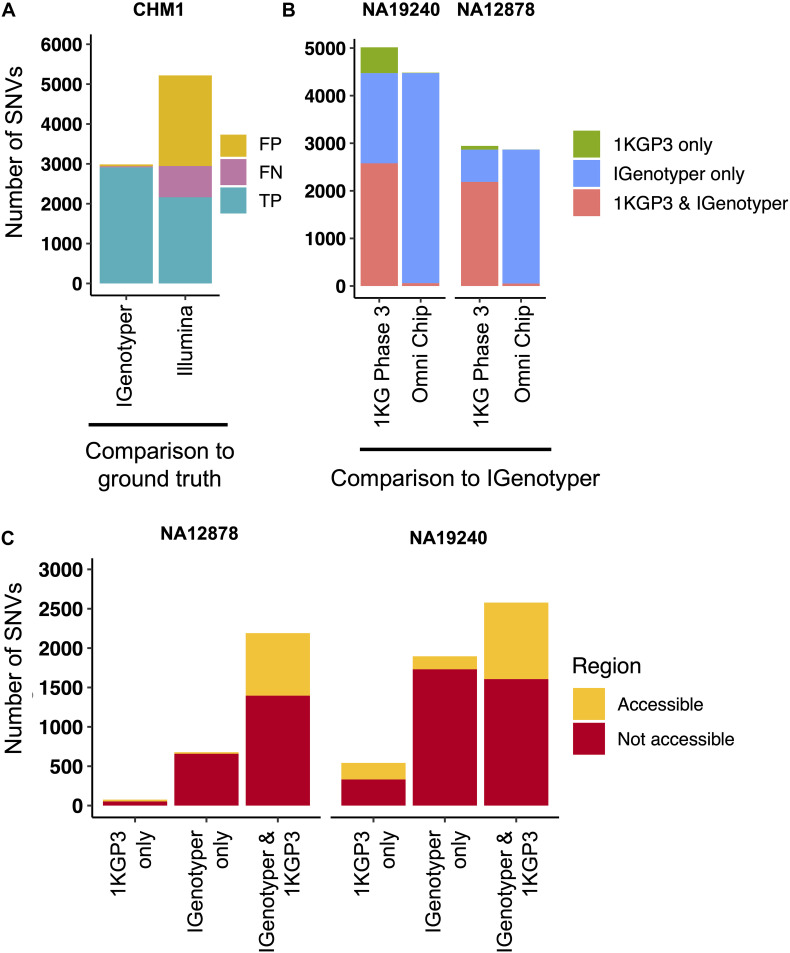
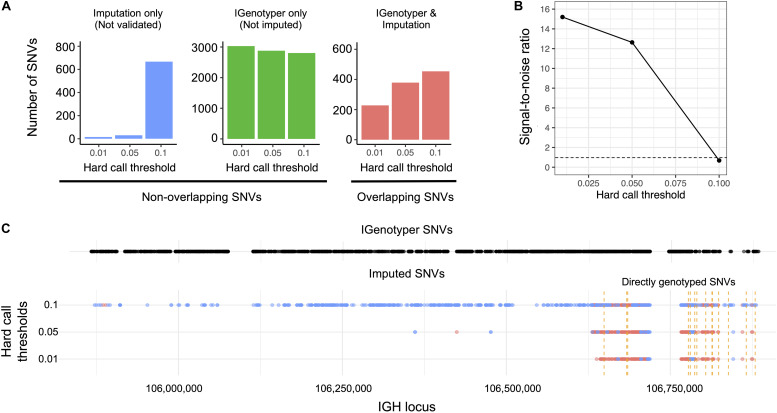
An incomplete ascertainment of genetic variation within the highly polymorphic immunoglobulin heavy chain locus (IGH) has hindered our ability to define genetic factors that influence antibody-mediated processes. Due to locus complexity, standard high-throughput approaches have failed to accurately and comprehensively capture IGH polymorphism. As a result, the locus has only been fully characterized two times, severely limiting our knowledge of human IGH diversity. Here, we combine targeted long-read sequencing with a novel bioinformatics tool, IGenotyper, to fully characterize IGH variation in a haplotype-specific manner. We apply this approach to eight human samples, including a haploid cell line and two mother-father-child trios, and demonstrate the ability to generate high-quality assemblies (>98% complete and >99% accurate), genotypes, and gene annotations, identifying 2 novel structural variants and 15 novel IGH alleles. We show multiplexing allows for scaling of the approach without impacting data quality, and that our genotype call sets are more accurate than short-read (>35% increase in true positives and >97% decrease in false-positives) and array/imputation-based datasets. This framework establishes a desperately needed foundation for leveraging IG genomic data to study population-level variation in antibody-mediated immunity, critical for bettering our understanding of disease risk, and responses to vaccines and therapeutics.
由于高度多态性的免疫球蛋白重链基因座(IGH)内遗传变异的不完全确定,我们定义影响抗体介导过程的遗传因素的能力受到了阻碍。由于基因座的复杂性,标准的高通量方法无法准确全面地捕捉IGH 多态性。因此,该基因座仅被完全描述了两次,严重限制了我们对人类 IGH 多样性的了解。在这里,我们将靶向长读测序与一种新的生物信息学工具 IGenotyper 相结合,以特定于单倍型的方式全面描述IGH 变异。我们将这种方法应用于八个人类样本,包括一个单倍体细胞系和两个母子父子三人组,并证明了生成高质量组装体(>98%完整和>99%准确)、基因型和基因注释的能力,确定了 2 个新的结构变体和 15 个新的 IGH 等位基因。我们表明,多重处理可以在不影响数据质量的情况下扩展该方法,并且我们的基因型调用集比短读(>35%的真阳性增加和>97%的假阳性减少)和基于阵列/插补的数据集更准确。该框架为利用 IG 基因组数据研究抗体介导免疫的群体水平变异奠定了迫切需要的基础,这对于更好地理解疾病风险以及对疫苗和治疗药物的反应至关重要。