Pacific Biosciences, Menlo Park, CA, USA.
Google Inc., Mountain View, CA, USA.
Nat Biotechnol. 2019 Oct;37(10):1155-1162. doi: 10.1038/s41587-019-0217-9. Epub 2019 Aug 12.
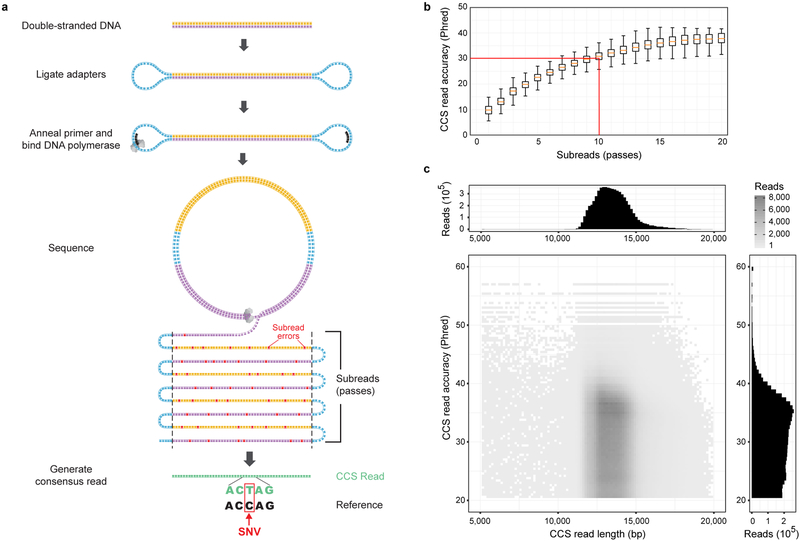
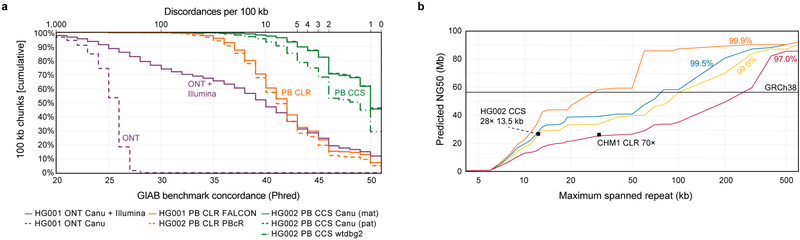
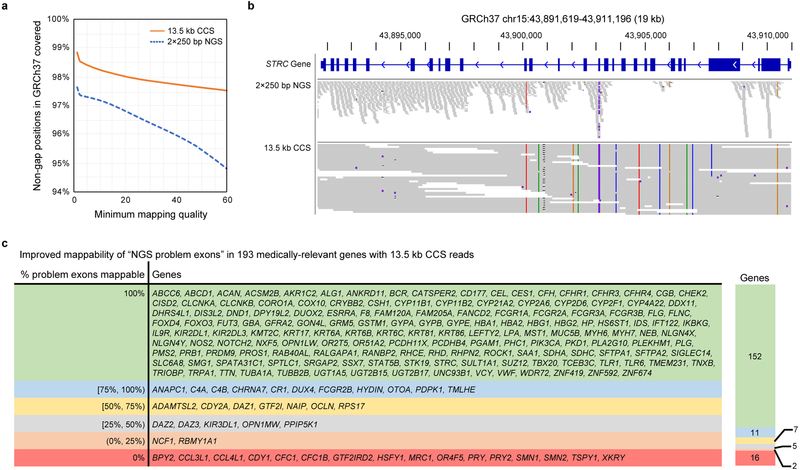
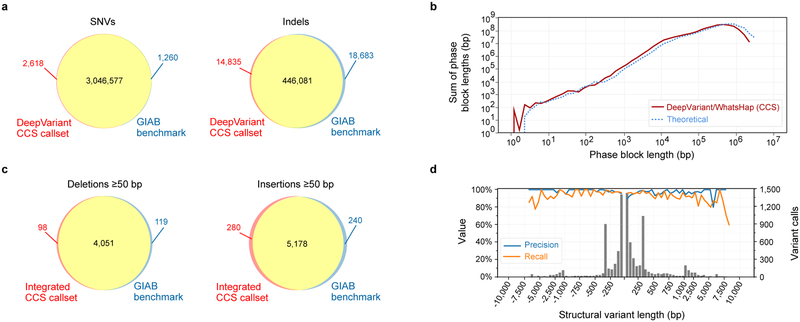
The DNA sequencing technologies in use today produce either highly accurate short reads or less-accurate long reads. We report the optimization of circular consensus sequencing (CCS) to improve the accuracy of single-molecule real-time (SMRT) sequencing (PacBio) and generate highly accurate (99.8%) long high-fidelity (HiFi) reads with an average length of 13.5 kilobases (kb). We applied our approach to sequence the well-characterized human HG002/NA24385 genome and obtained precision and recall rates of at least 99.91% for single-nucleotide variants (SNVs), 95.98% for insertions and deletions <50 bp (indels) and 95.99% for structural variants. Our CCS method matches or exceeds the ability of short-read sequencing to detect small variants and structural variants. We estimate that 2,434 discordances are correctable mistakes in the 'genome in a bottle' (GIAB) benchmark set. Nearly all (99.64%) variants can be phased into haplotypes, further improving variant detection. De novo genome assembly using CCS reads alone produced a contiguous and accurate genome with a contig N50 of >15 megabases (Mb) and concordance of 99.997%, substantially outperforming assembly with less-accurate long reads.
目前使用的 DNA 测序技术要么产生高度准确的短读长,要么产生不太准确的长读长。我们报告了对环形一致性测序(CCS)的优化,以提高单分子实时(SMRT)测序(PacBio)的准确性,并生成高度准确(99.8%)的长高保真度(HiFi)读长,平均长度为 13.5 千碱基(kb)。我们应用我们的方法对特征明确的人类 HG002/NA24385 基因组进行测序,并获得至少 99.91%的单核苷酸变异(SNV)、95.98%的<50 bp 插入和缺失(indels)和 95.99%的结构变异的精度和召回率。我们的 CCS 方法与短读长测序检测小变异和结构变异的能力相匹配或超过。我们估计在“瓶中基因组”(GIAB)基准集中,2434 个不一致是可纠正的错误。几乎所有(99.64%)的变体都可以被相位成单倍型,进一步提高了变体检测的准确性。单独使用 CCS 读取进行从头基因组组装生成了一个连续且准确的基因组,其 contig N50 大于 15 兆碱基(Mb),一致性为 99.997%,明显优于使用不太准确的长读长进行的组装。