Physikalische Chemie III, Technische Universität Dortmund, Otto-Hahn-Str. 4a, 44227, Dortmund, Germany.
R&D Integrated Drug Discovery, Sanofi-Aventis Deutschland GmbH, 65926, Frankfurt am Main, Germany.
J Comput Aided Mol Des. 2021 Apr;35(4):453-472. doi: 10.1007/s10822-020-00347-5. Epub 2020 Oct 20.
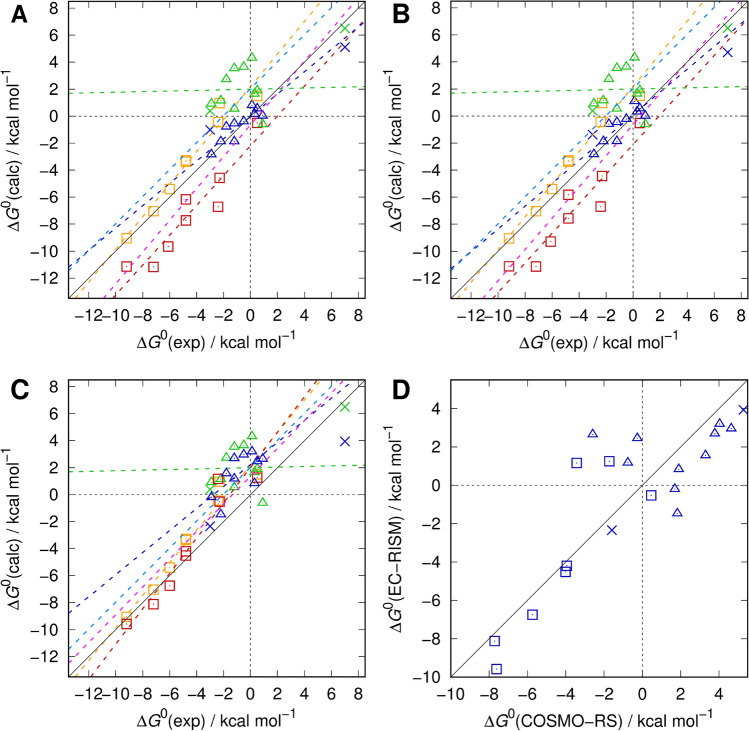
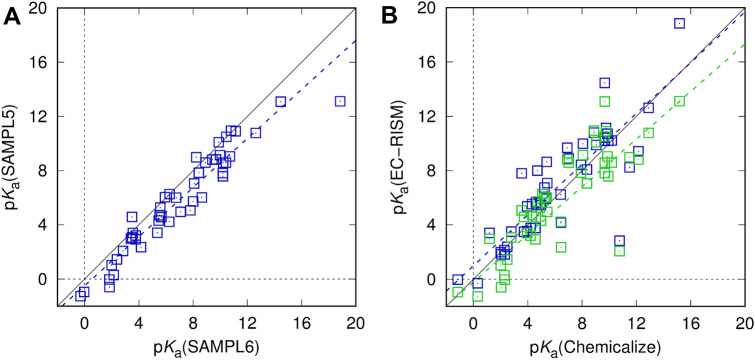
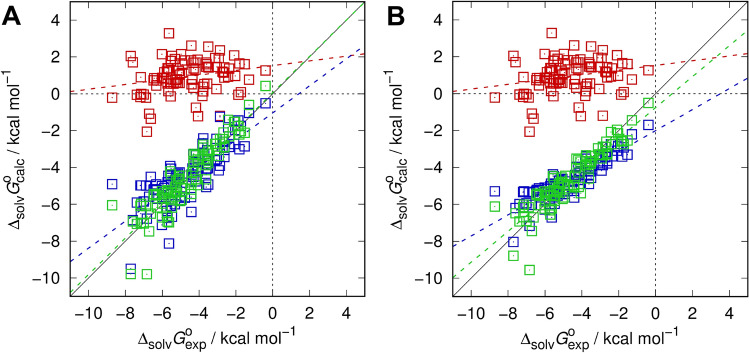
Joint academic-industrial projects supporting drug discovery are frequently pursued to deploy and benchmark cutting-edge methodical developments from academia in a real-world industrial environment at different scales. The dimensionality of tasks ranges from small molecule physicochemical property assessment over protein-ligand interaction up to statistical analyses of biological data. This way, method development and usability both benefit from insights gained at both ends, when predictiveness and readiness of novel approaches are confirmed, but the pharmaceutical drug makers get early access to novel tools for the quality of drug products and benefit of patients. Quantum-mechanical and simulation methods particularly fall into this group of methods, as they require skills and expense in their development but also significant resources in their application, thus are comparatively slowly dripping into the realm of industrial use. Nevertheless, these physics-based methods are becoming more and more useful. Starting with a general overview of these and in particular quantum-mechanical methods for drug discovery we review a decade-long and ongoing collaboration between Sanofi and the Kast group focused on the application of the embedded cluster reference interaction site model (EC-RISM), a solvation model for quantum chemistry, to study small molecule chemistry in the context of joint participation in several SAMPL (Statistical Assessment of Modeling of Proteins and Ligands) blind prediction challenges. Starting with early application to tautomer equilibria in water (SAMPL2) the methodology was further developed to allow for challenge contributions related to predictions of distribution coefficients (SAMPL5) and acidity constants (SAMPL6) over the years. Particular emphasis is put on a frequently overlooked aspect of measuring the quality of models, namely the retrospective analysis of earlier datasets and predictions in light of more recent and advanced developments. We therefore demonstrate the performance of the current methodical state of the art as developed and optimized for the SAMPL6 pK and octanol-water log P challenges when re-applied to the earlier SAMPL5 cyclohexane-water log D and SAMPL2 tautomer equilibria datasets. Systematic improvement is not consistently found throughout despite the similarity of the problem class, i.e. protonation reactions and phase distribution. Hence, it is possible to learn about hidden bias in model assessment, as results derived from more elaborate methods do not necessarily improve quantitative agreement. This indicates the role of chance or coincidence for model development on the one hand which allows for the identification of systematic error and opportunities toward improvement and reveals possible sources of experimental uncertainty on the other. These insights are particularly useful for further academia-industry collaborations, as both partners are then enabled to optimize both the computational and experimental settings for data generation.
产学研合作项目经常被用于将学术领域的前沿方法学发展应用于现实工业环境中,并在不同规模下进行基准测试。任务的维度从小分子的物理化学性质评估、蛋白质-配体相互作用到生物数据的统计分析都有涉及。这样一来,当新方法的预测能力和准备程度得到验证时,方法的开发和可用性都可以从双方获得的见解中受益,制药商也可以尽早获得新药产品质量和患者受益的新工具。量子力学和模拟方法尤其属于这类方法,因为它们在开发过程中需要技能和费用,并且在应用中也需要大量资源,因此相对较慢地融入工业应用领域。尽管如此,这些基于物理的方法变得越来越有用。我们从对这些方法,特别是用于药物发现的量子力学方法的概述开始,回顾了赛诺菲(Sanofi)和 Kast 小组之间长达十年的合作,重点是应用嵌入簇参考相互作用点模型(EC-RISM),一种用于量子化学的溶剂化模型,来研究小分子化学,合作内容包括联合参与几个 SAMPL(蛋白质和配体建模的统计评估)盲测挑战。该方法最早应用于水中互变异构平衡(SAMPL2),随后多年来进一步发展,以允许对与预测分配系数(SAMPL5)和酸度常数(SAMPL6)相关的挑战做出贡献。特别强调的是,在回顾性分析早期数据集和预测时,经常忽略了衡量模型质量的一个方面,即根据最近和更先进的发展来分析。因此,我们展示了当前方法学最先进的性能,这些性能是针对 SAMPL6 pK 和辛醇-水 log P 挑战开发和优化的,当重新应用于早期的 SAMPL5 环己烷-水 log D 和 SAMPL2 互变异构平衡数据集时,性能得到了显著提高。尽管问题类(质子化反应和相分布)相似,但并未始终发现系统改进。因此,可以了解模型评估中的隐藏偏差,因为来自更复杂方法的结果不一定能提高定量一致性。这表明模型开发中的机会或巧合作用一方面允许识别系统误差和改进机会,并揭示实验不确定性的可能来源。这些见解对于进一步的产学研合作特别有用,因为双方都能够优化计算和实验设置以生成数据。