Schools of Basic Medicine and Pharmacy, Weifang Medical University, 7166 Baotong West Street, Weifang, 261053 Shandong Province, China.
Biomed Res Int. 2020 Oct 6;2020:1371632. doi: 10.1155/2020/1371632. eCollection 2020.
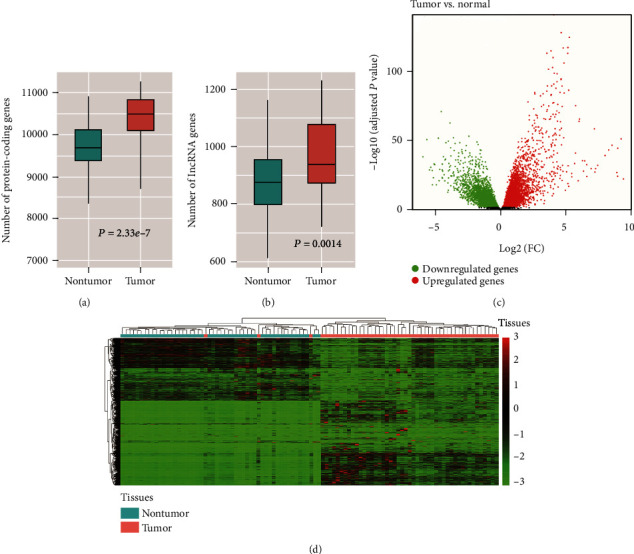
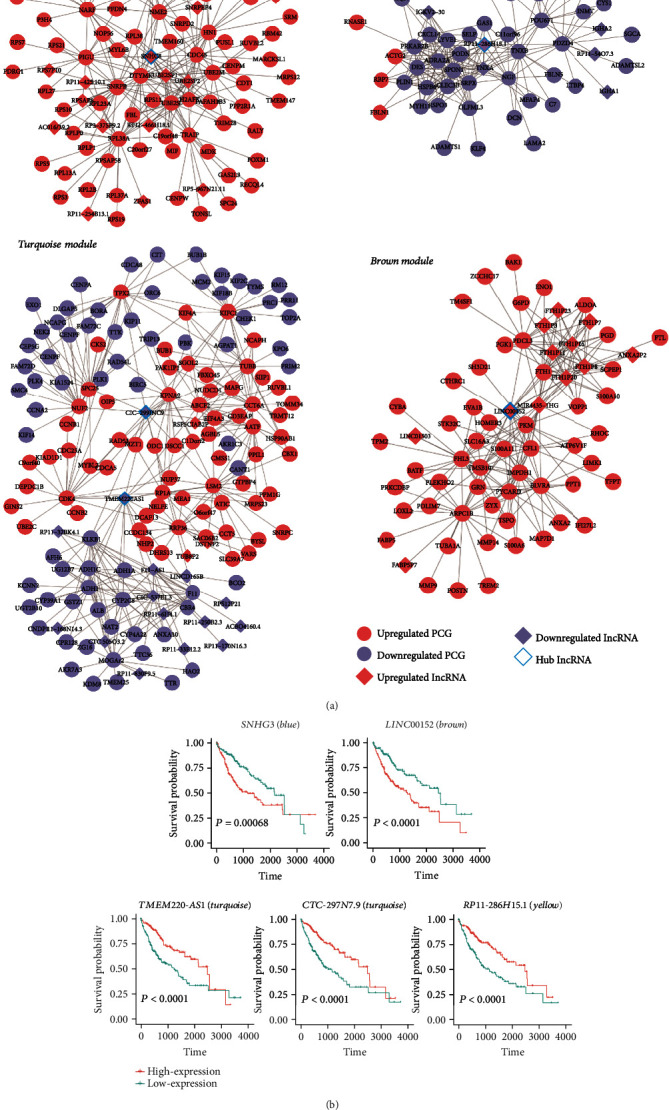
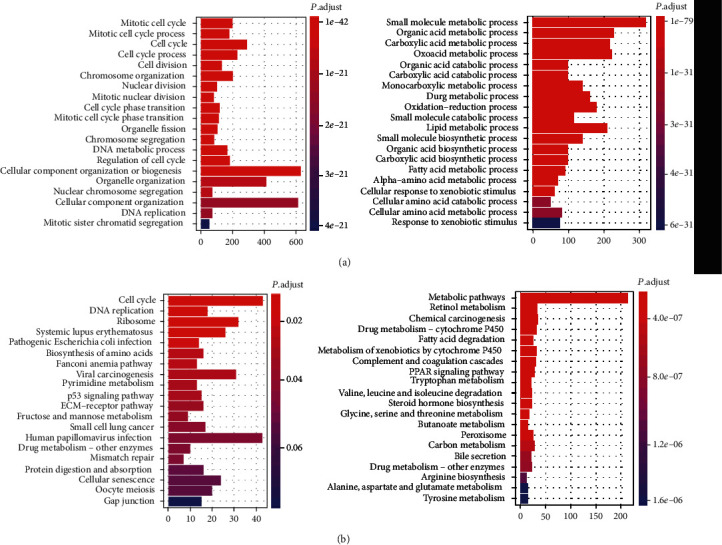
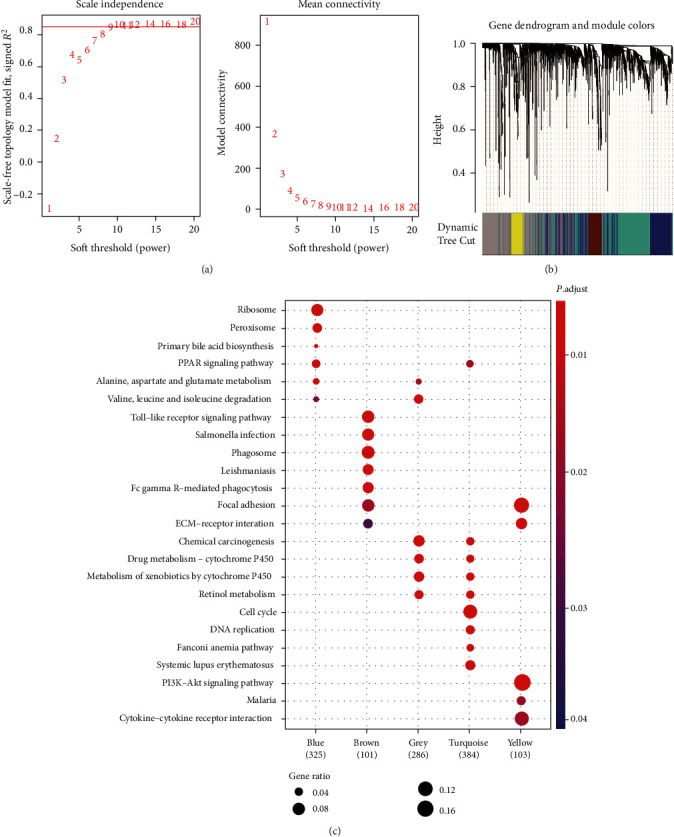
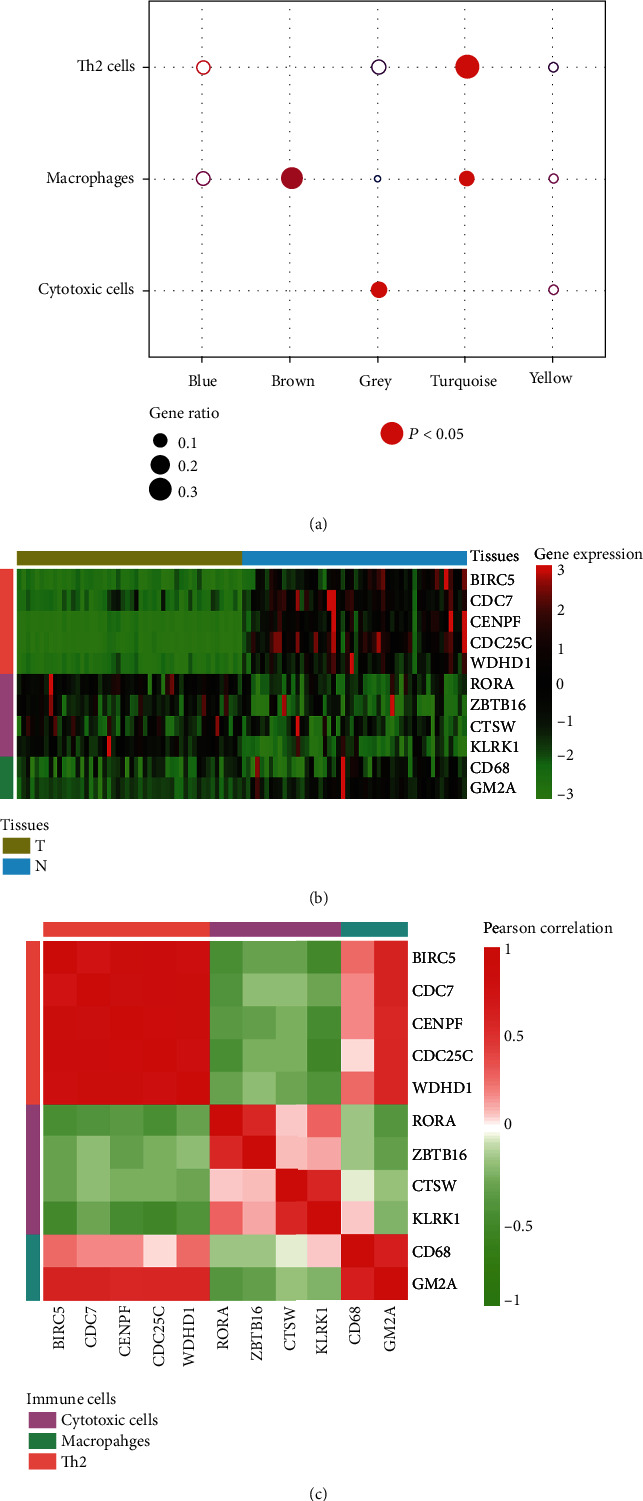
Hepatocellular carcinoma (HCC) is a primary liver cancer associated with a growing incidence and extremely high mortality. However, the pathogenic mechanism is still not fully understood. In the present study, we identified 1,631 upregulated and 1,515 downregulated genes and found that cell cycle and metabolism-related pathways or biological processes highly dysregulated in HCC. To assess the biological importance of these DEGs, we carried out weighted gene coexpression network analysis (WGCNA) to identify the functional modules potentially involved in HCC pathogenesis or progression. The five modules were detected with Dynamic Tree Cut algorithm, and GO enrichment analysis revealed that these modules exhibited different biological processes or signaling pathways, such as metabolism-related pathways, cell proliferation-related pathways, and molecules in tumor microenvironment. Moreover, we also observed two immune cells, namely, cytotoxic cells and macrophage enriched in modules grey and brown, respectively, while T helper cell-2 (Th2) was enriched in module turquoise. Among the WGCNA network, four hub long noncoding RNAs (lncRNAs) were identified to be associated with HCC prognostic outcomes, suggesting that coexpression network analysis could uncover lncRNAs with functional importance, which may be associated with prognostic outcomes of HCC patients. In summary, this study demonstrated that network-based analysis could identify some functional modules and some hub-lncRNAs, which may be critical for HCC pathogenesis or progression.
肝细胞癌(HCC)是一种原发性肝癌,其发病率不断增加,死亡率极高。然而,其发病机制仍不完全清楚。在本研究中,我们鉴定了 1631 个上调基因和 1515 个下调基因,发现细胞周期和代谢相关途径或生物过程在 HCC 中高度失调。为了评估这些差异表达基因(DEGs)的生物学重要性,我们进行了加权基因共表达网络分析(WGCNA),以鉴定可能参与 HCC 发病机制或进展的功能模块。使用动态树切割算法检测到五个模块,GO 富集分析表明这些模块表现出不同的生物学过程或信号通路,如代谢相关途径、细胞增殖相关途径和肿瘤微环境中的分子。此外,我们还观察到两个免疫细胞,即细胞毒性细胞和巨噬细胞分别富集在模块灰色和棕色中,而 Th2 细胞在模块绿松石中富集。在 WGCNA 网络中,鉴定出四个与 HCC 预后结局相关的关键长非编码 RNA(lncRNA),提示共表达网络分析可以揭示具有功能重要性的 lncRNA,可能与 HCC 患者的预后结局相关。总之,本研究表明,基于网络的分析可以识别一些功能模块和一些关键的 lncRNA,这些可能对 HCC 的发病机制或进展至关重要。