Department of Immunology and Infectious Diseases, Harvard T.H. Chan School of Public Health, Harvard T.H. Chan School of Public Health, 665 Huntington Ave, Boston, MA, 02115, USA.
Broad Institute, Cambridge, MA, USA.
Malar J. 2020 Oct 23;19(1):379. doi: 10.1186/s12936-020-03439-7.
With increasing interest in eliminating malaria from the Caribbean region, Haiti is one of the two countries on the island of Hispaniola with continued malaria transmission. While the Haitian population remains at risk for malaria, there are a limited number of cases annually, making conventional epidemiological measures such as case incidence and prevalence of potentially limited value for fine-scale resolution of transmission patterns and trends. In this context, genetic signatures may be useful for the identification and characterization of the Plasmodium falciparum parasite population in order to identify foci of transmission, detect outbreaks, and track parasite movement to potentially inform malaria control and elimination strategies.
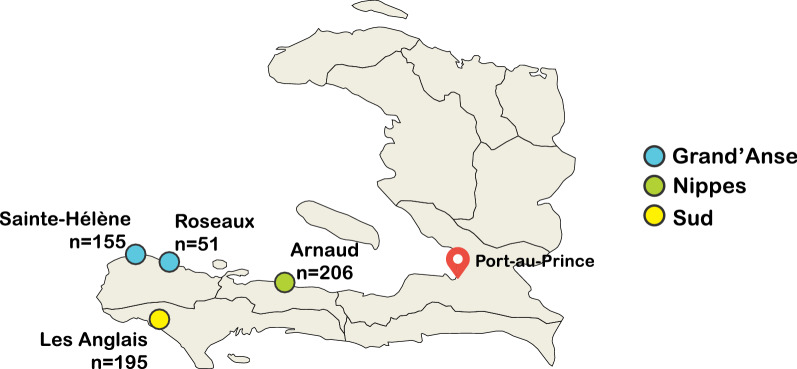
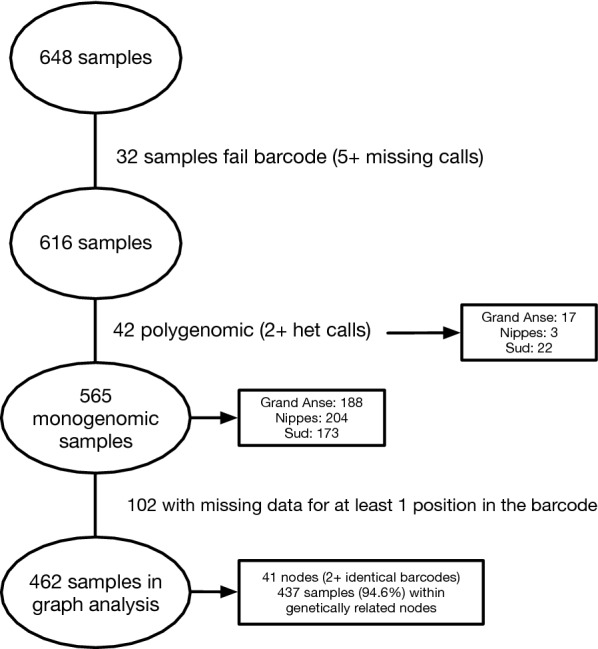
This study evaluated the genetic signals based on analysis of 21 single-nucleotide polymorphisms (SNPs) from 462 monogenomic (single-genome) P. falciparum DNA samples extracted from dried blood spots collected from malaria-positive patients reporting to health facilities in three southwestern Haitian departments (Nippes, Grand'Anse, and Sud) in 2016.
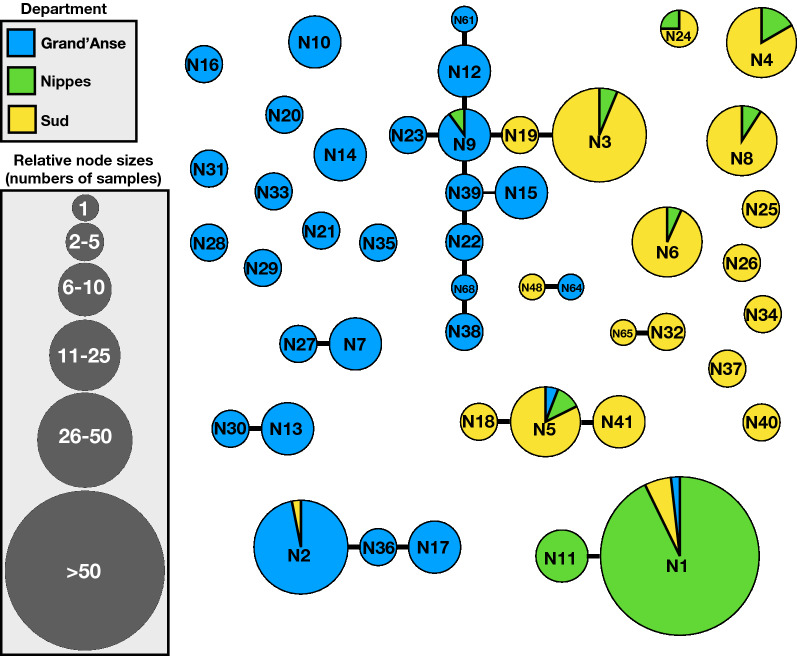
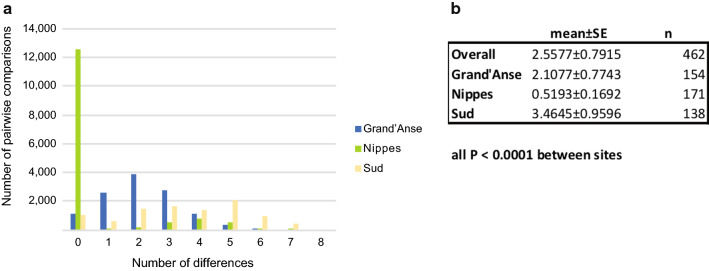
Assessment of the parasite genetic relatedness revealed evidence of clonal expansion within Nippes and the exchange of parasite lineages between Nippes, Sud, and Grand'Anse. Furthermore, 437 of the 462 samples shared high levels of genetic similarity-at least 20 of 21 SNPS-with at least one other sample in the dataset.
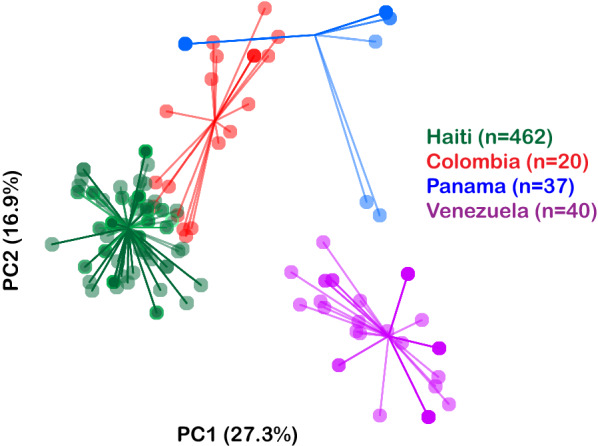
These results revealed patterns of relatedness suggestive of the repeated recombination of a limited number of founding parasite types without significant outcrossing. These genetic signals offer clues to the underlying relatedness of parasite populations and may be useful for the identification of the foci of transmission and tracking of parasite movement in Haiti for malaria elimination.
随着人们对从加勒比地区消除疟疾的兴趣日益浓厚,海地是伊斯帕尼奥拉岛上仍有疟疾传播的两个国家之一。虽然海地人口仍然面临疟疾风险,但每年的病例数量有限,因此传统的流行病学措施(如病例发病率和患病率)对于精细解析传播模式和趋势可能具有一定的局限性。在这种情况下,遗传特征可能有助于鉴定和描述恶性疟原虫寄生虫群体,以确定传播焦点,发现疫情,并跟踪寄生虫的移动,从而为疟疾控制和消除策略提供信息。
本研究评估了基于对 21 个单核苷酸多态性(SNP)的遗传信号,分析了从 2016 年从报告有疟疾的卫生机构就诊的 3 个海地西南部省份(尼普斯、大湾和南部)的 462 个恶性疟原虫单基因组 DNA 样本中提取的 462 个单基因组(单个基因组)P. falciparum DNA 样本。
寄生虫遗传相关性评估显示,尼普斯内部存在克隆扩张的证据,以及尼普斯、南部和大湾之间寄生虫谱系的交换。此外,462 个样本中的 437 个样本与数据集中的至少一个其他样本具有至少 21 个 SNP 中的 20 个 SNP 的高度遗传相似性。
这些结果揭示了相关性模式,提示有限数量的原始寄生虫类型反复重组,而没有明显的杂交。这些遗传特征为寄生虫种群的潜在相关性提供了线索,可能有助于确定海地的传播焦点,并跟踪寄生虫的移动,以实现消除疟疾的目标。