McDonald Institute for Archaeological Research, University of Cambridge, Cambridge, UK.
Department of Human Genetics, Katholieke Universiteit Leuven, Herestraat 49 - box 602, 3000, Leuven, Belgium.
Sci Rep. 2020 Oct 29;10(1):18542. doi: 10.1038/s41598-020-75387-w.
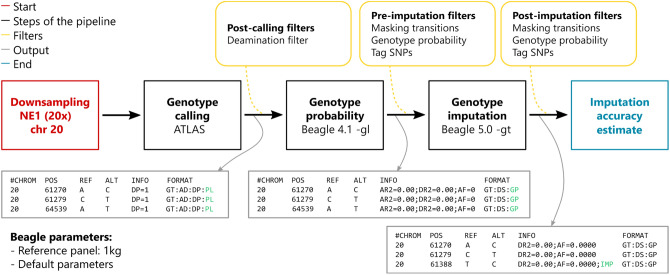
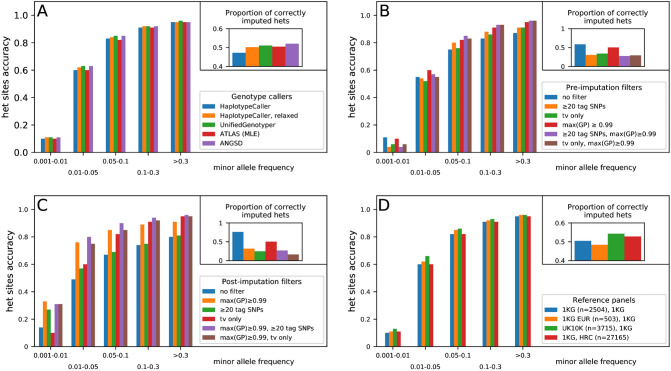
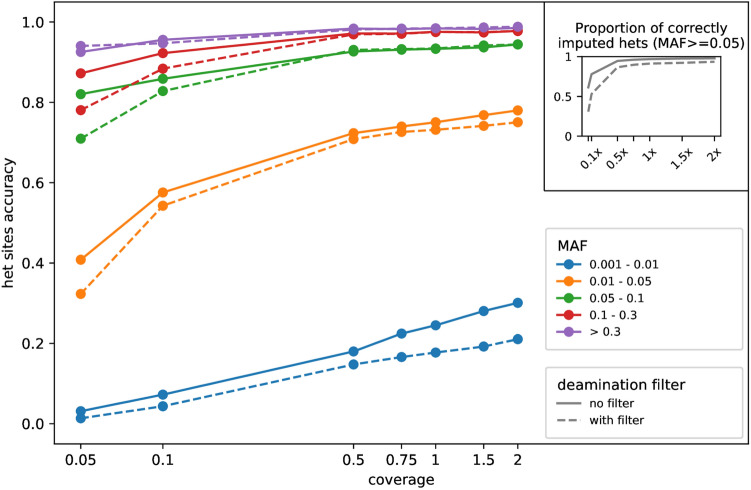
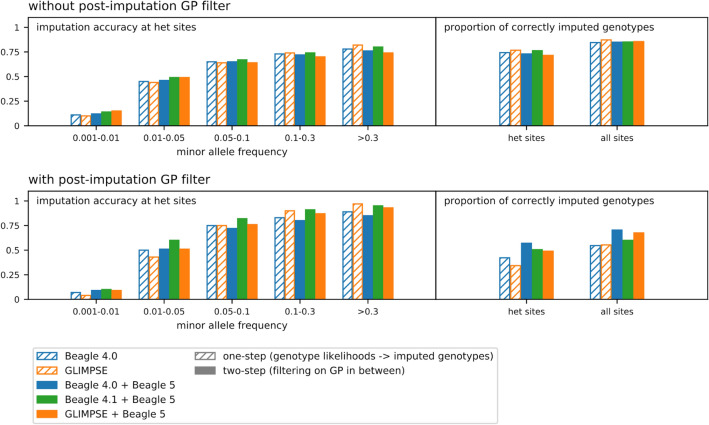
Although ancient DNA data have become increasingly more important in studies about past populations, it is often not feasible or practical to obtain high coverage genomes from poorly preserved samples. While methods of accurate genotype imputation from > 1 × coverage data have recently become a routine, a large proportion of ancient samples remain unusable for downstream analyses due to their low coverage. Here, we evaluate a two-step pipeline for the imputation of common variants in ancient genomes at 0.05-1 × coverage. We use the genotype likelihood input mode in Beagle and filter for confident genotypes as the input to impute missing genotypes. This procedure, when tested on ancient genomes, outperforms a single-step imputation from genotype likelihoods, suggesting that current genotype callers do not fully account for errors in ancient sequences and additional quality controls can be beneficial. We compared the effect of various genotype likelihood calling methods, post-calling, pre-imputation and post-imputation filters, different reference panels, as well as different imputation tools. In a Neolithic Hungarian genome, we obtain ~ 90% imputation accuracy for heterozygous common variants at coverage 0.05 × and > 97% accuracy at coverage 0.5 ×. We show that imputation can mitigate, though not eliminate reference bias in ultra-low coverage ancient genomes.
尽管古 DNA 数据在过去人群的研究中变得越来越重要,但从保存不佳的样本中获得高覆盖率基因组通常是不可行或不实际的。虽然最近已经可以从超过 1x 的覆盖数据中进行准确的基因型推断的方法已经成为常规方法,但由于其覆盖率低,仍有很大一部分古代样本无法用于下游分析。在这里,我们评估了一种两步法在 0.05-1x 覆盖率下对古代基因组中的常见变体进行推断的方法。我们在 Beagle 中使用基因型似然输入模式,并过滤出置信基因型作为输入来推断缺失的基因型。当在古代基因组上进行测试时,该程序优于从基因型似然的单步推断,这表明当前的基因型呼叫器没有充分考虑到古代序列中的错误,并且可以进行额外的质量控制。我们比较了不同的基因型似然呼叫方法、呼叫后、预推断和后推断过滤、不同的参考面板以及不同的推断工具的效果。在新石器时代的匈牙利基因组中,我们在覆盖率为 0.05x 时获得了~90%的杂合常见变体的推断准确性,在覆盖率为 0.5x 时获得了>97%的准确性。我们表明,推断可以减轻,但不能消除超低覆盖率古代基因组中的参考偏差。