Jeon Yoon Sun, Cha Jae Kook, Choi Seong Ho, Lee Ji Hyun, Lee Jung Seok
Department of Periodontology, Research Institute of Periodontal Regeneration, Yonsei University College of Dentistry, Seoul, Korea.
Department of Clinical Pharmacology and Therapeutics, Kyung Hee University College of Medicine, Seoul, Korea.
J Periodontal Implant Sci. 2020 Oct;50(5):313-326. doi: 10.5051/jpis.1905460273.
This study was conducted to analyze specific RNA expression profiles in gingival tissue and saliva samples in periodontitis patients and healthy individuals, and to determine their correlations in light of the potential use of microarray-based analyses of saliva samples as a periodontal monitoring tool.
Gingival tissue biopsies and saliva samples from 22 patients (12 with severe periodontitis and 10 with a healthy periodontium) were analyzed using transcriptomic microarray analysis. Differential gene expression was assessed, and pathway and clustering analyses were conducted for the samples. The correlations between the results for the gingival tissue and saliva samples were analyzed at both the gene and pathway levels.
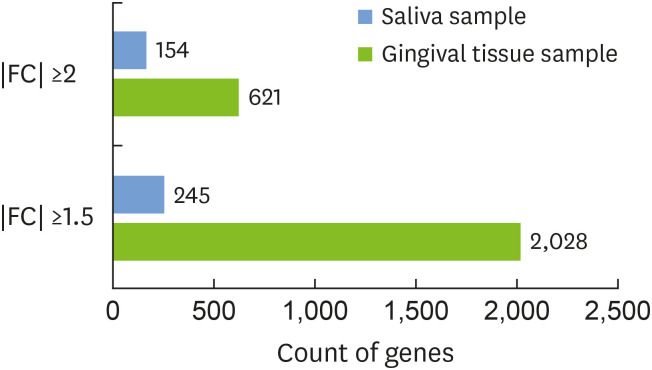
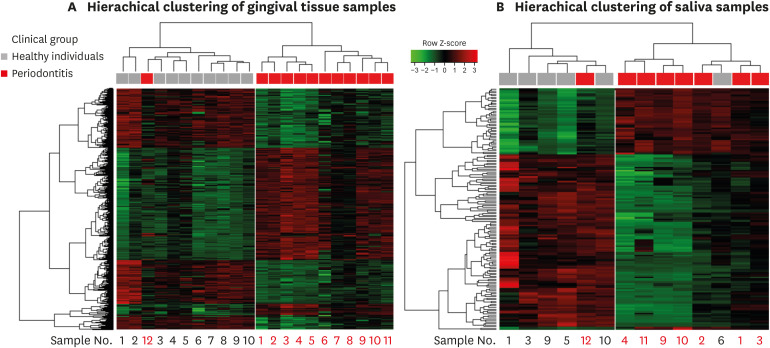
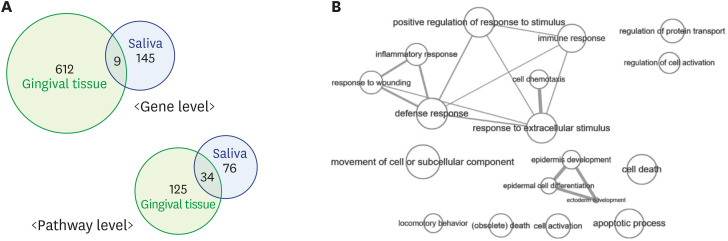
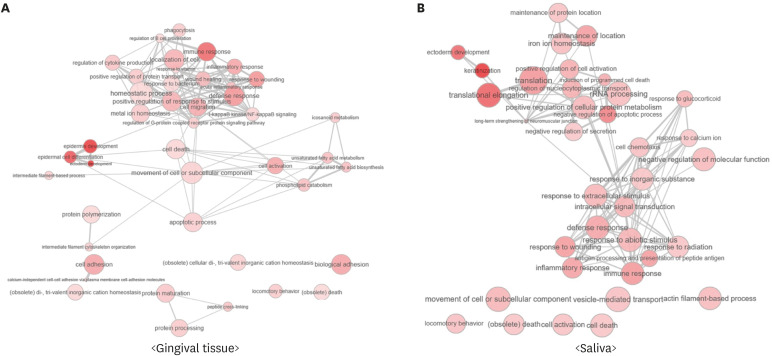
There were 621 differentially expressed genes (DEGs; 320 upregulated and 301 downregulated) in the gingival tissue samples of the periodontitis group, and 154 DEGs (44 upregulated and 110 downregulated) in the saliva samples. Nine of these genes overlapped between the sample types. The periodontitis patients formed a distinct cluster group based on gene expression profiles for both the tissue and saliva samples. Database for Annotation, Visualization and Integrated Discovery analysis revealed 159 enriched pathways from the tissue samples of the periodontitis patients, as well as 110 enriched pathways In the saliva samples. Thirty-four pathways overlapped between the sample types.
The present results indicate the possibility of using the salivary transcriptome to distinguish periodontitis patients from healthy individuals. Further work is required to enhance the extraction of available RNA from saliva samples.
本研究旨在分析牙周炎患者和健康个体牙龈组织及唾液样本中的特定RNA表达谱,并鉴于基于微阵列分析唾液样本作为牙周监测工具的潜在用途,确定它们之间的相关性。
对22例患者(12例重度牙周炎患者和10例牙周健康者)的牙龈组织活检样本和唾液样本进行转录组微阵列分析。评估差异基因表达,并对样本进行通路和聚类分析。在基因和通路水平上分析牙龈组织和唾液样本结果之间的相关性。
牙周炎组牙龈组织样本中有621个差异表达基因(DEGs;320个上调,301个下调),唾液样本中有154个DEGs(44个上调,110个下调)。其中9个基因在样本类型之间重叠。基于组织和唾液样本的基因表达谱,牙周炎患者形成了一个独特的聚类组。注释、可视化和综合发现数据库分析显示,牙周炎患者组织样本中有159条富集通路,唾液样本中有110条富集通路。样本类型之间有34条通路重叠。
目前的结果表明,利用唾液转录组区分牙周炎患者和健康个体是有可能的。需要进一步开展工作以提高从唾液样本中提取可用RNA的效率。