Department of Chemistry, Duke University, Durham, NC, 27710, USA.
Department of Biochemistry, Duke University School of Medicine, Durham, NC, 27710, USA.
Nat Commun. 2020 Nov 2;11(1):5531. doi: 10.1038/s41467-020-19371-y.
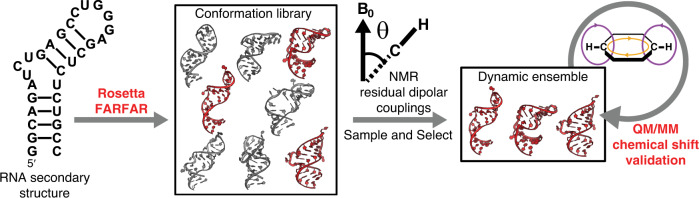
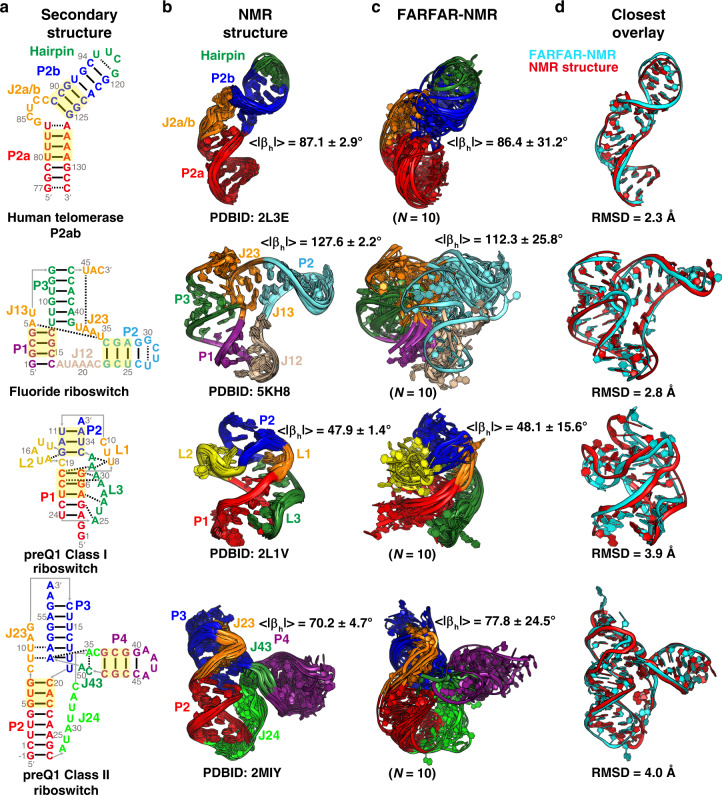
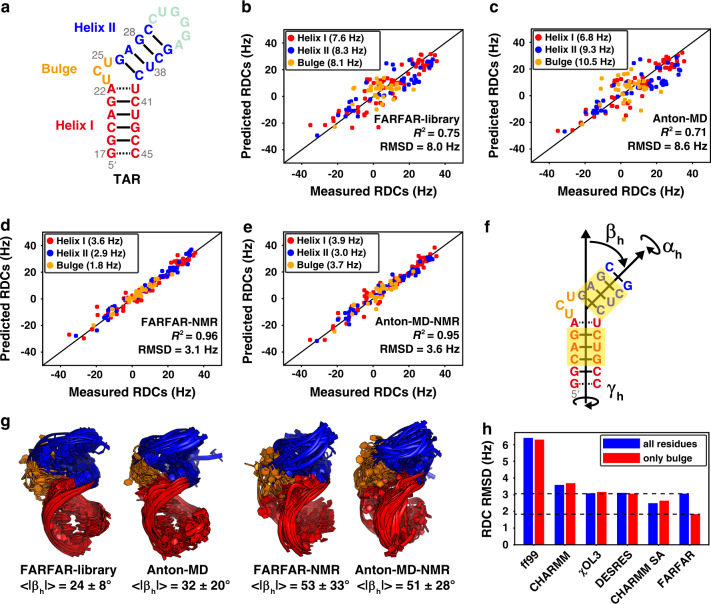
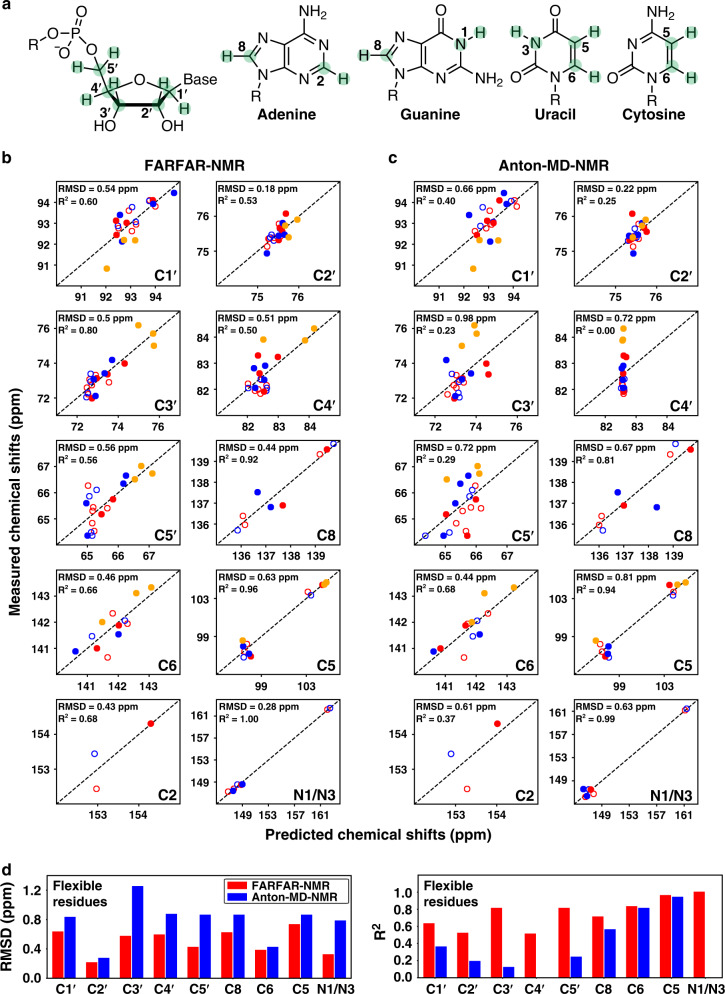
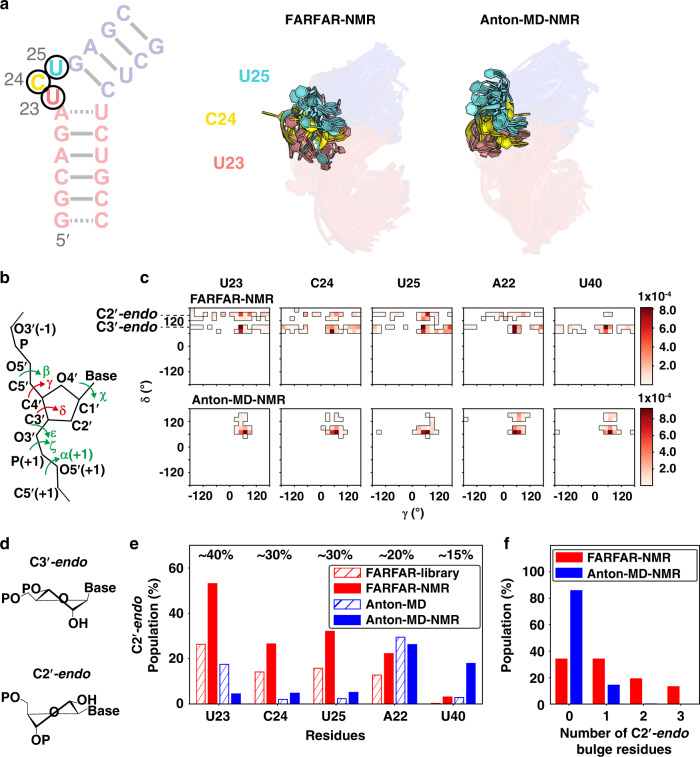
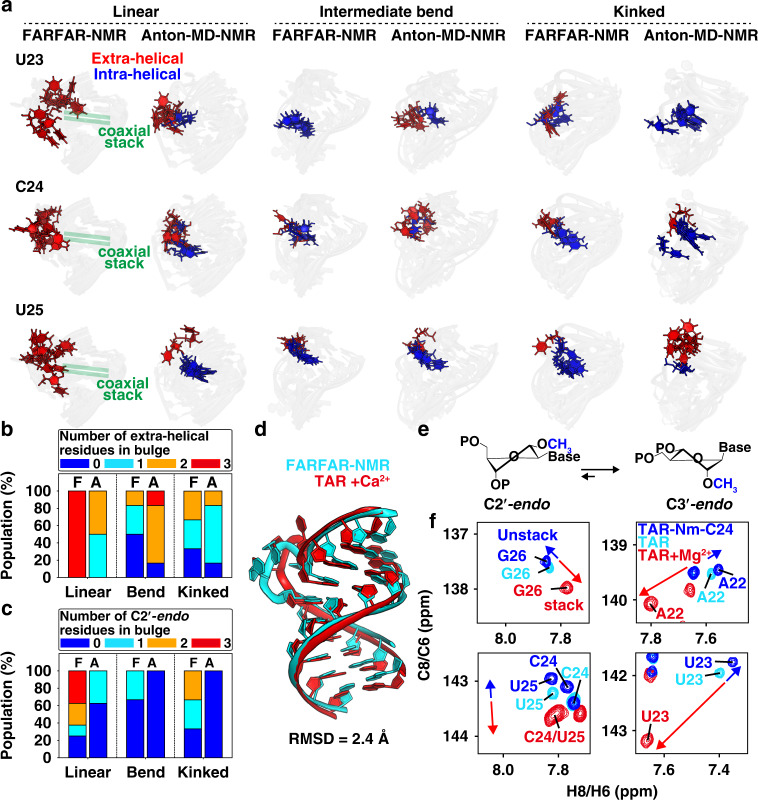
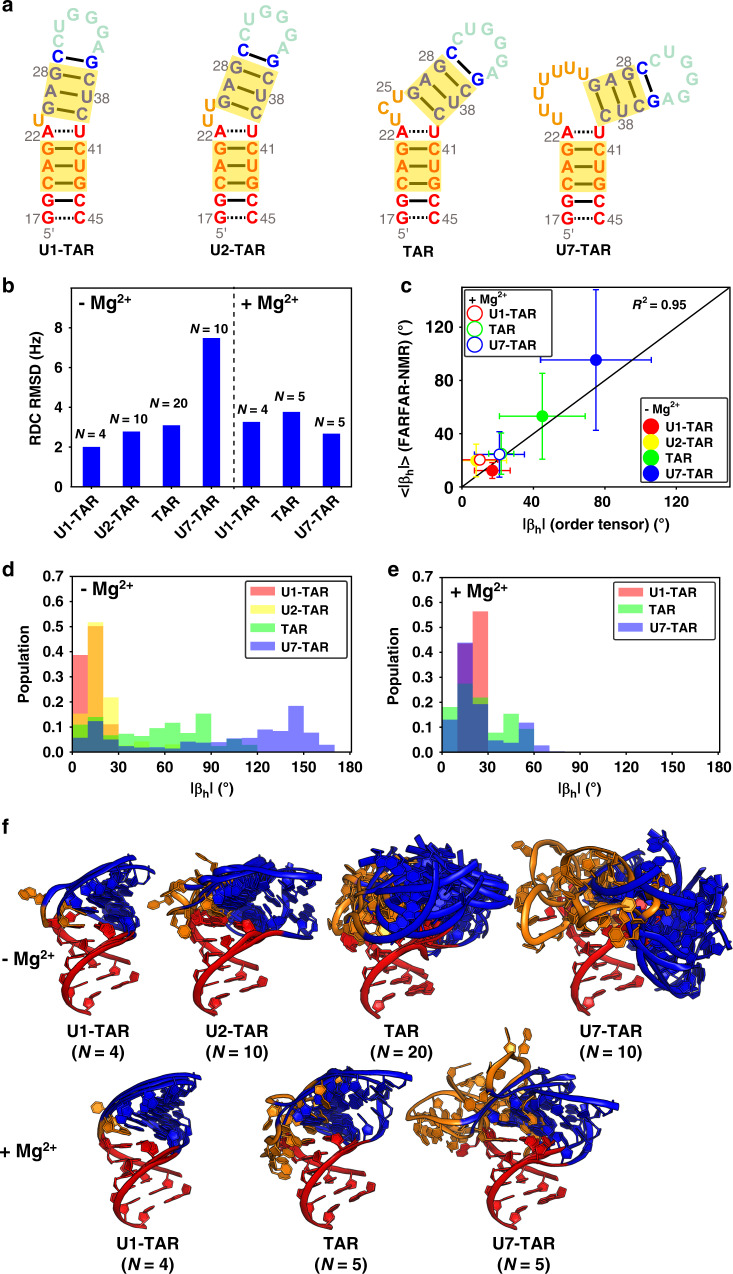
Biomolecules form dynamic ensembles of many inter-converting conformations which are key for understanding how they fold and function. However, determining ensembles is challenging because the information required to specify atomic structures for thousands of conformations far exceeds that of experimental measurements. We addressed this data gap and dramatically simplified and accelerated RNA ensemble determination by using structure prediction tools that leverage the growing database of RNA structures to generate a conformation library. Refinement of this library with NMR residual dipolar couplings provided an atomistic ensemble model for HIV-1 TAR, and the model accuracy was independently supported by comparisons to quantum-mechanical calculations of NMR chemical shifts, comparison to a crystal structure of a substate, and through designed ensemble redistribution via atomic mutagenesis. Applications to TAR bulge variants and more complex tertiary RNAs support the generality of this approach and the potential to make the determination of atomic-resolution RNA ensembles routine.
生物分子形成了由多种相互转化构象组成的动态集合,这对于理解它们如何折叠和发挥功能至关重要。然而,确定这些集合具有挑战性,因为指定数千种构象的原子结构所需的信息远远超出了实验测量的范围。我们通过使用结构预测工具解决了这个数据缺口,这些工具利用日益增长的 RNA 结构数据库来生成构象库,从而极大地简化和加速了 RNA 集合的确定。利用 NMR 残差偶极耦合对该文库进行细化,为 HIV-1 TAR 提供了一个原子集合模型,该模型的准确性通过与 NMR 化学位移的量子力学计算进行比较、与亚态晶体结构进行比较以及通过原子诱变进行设计的集合再分配得到了独立的支持。对 TAR 突环变体和更复杂的三级 RNA 的应用支持了这种方法的通用性,并有可能使原子分辨率 RNA 集合的确定成为常规。