Global Discovery Chemistry, Computer-Aided Drug Discovery, Novartis Institutes for BioMedical Research, Cambridge, Massachusetts, United States of America.
PLoS One. 2020 Nov 4;15(11):e0234946. doi: 10.1371/journal.pone.0234946. eCollection 2020.
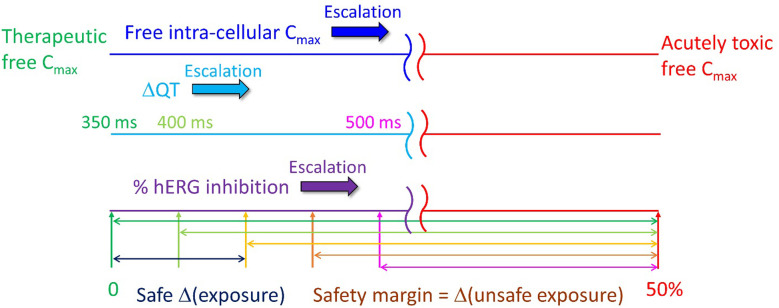
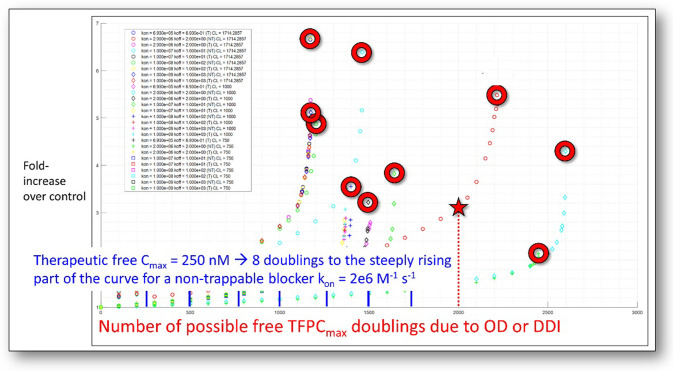
The human ether-a-go-go-related voltage-gated cardiac ion channel (commonly known as hERG) conducts the rapid outward repolarizing potassium current in cardiomyocytes (IKr). Inadvertent blockade of this channel by drug-like molecules represents a key challenge in pharmaceutical R&D due to frequent overlap between the structure-activity relationships of hERG and many primary targets. Building on our previous work, together with recent cryo-EM structures of hERG, we set about to better understand the energetic and structural basis of promiscuous blocker-hERG binding in the context of Biodynamics theory. We propose a two-step blocker binding process consisting of: The initial capture step: diffusion of a single fully solvated blocker copy into a large cavity lined by the intra-cellular cyclic nucleotide binding homology domain (CNBHD). Occupation of this cavity is a necessary but insufficient condition for ion current disruption.The IKr disruption step: translocation of the captured blocker along the channel axis, such that: The head group, consisting of a quasi-rod-shaped moiety, projects into the open pore, accompanied by partial de-solvation of the binding interface.One tail moiety packs along a kink between the S6 helix and proximal C-linker helix adjacent to the intra-cellular entrance of the pore, likewise accompanied by mutual de-solvation of the binding interface (noting that the association barrier is comprised largely of the total head + tail group de-solvation cost).Blockers containing a highly planar moiety that projects into a putative constriction zone within the closed channel become trapped upon closing, as do blockers terminating prior to this region.A single captured blocker copy may conceivably associate and dissociate to/from the pore many times before exiting the CNBHD cavity. Lastly, we highlight possible flaws in the current hERG safety index (SI), and propose an alternate in vivo-relevant strategy factoring in: Benefit/risk.The predicted arrhythmogenic fractional hERG occupancy (based on action potential (AP) simulations of the undiseased human ventricular cardiomyocyte).Alteration of the safety threshold due to underlying disease.Risk of exposure escalation toward the predicted arrhythmic limit due to patient-to-patient pharmacokinetic (PK) variability, drug-drug interactions, overdose, and use for off-label indications in which the hERG safety parameters may differ from their on-label counterparts.
人类 Ether-A-Go-Go 相关电压门控心肌离子通道(通常称为 hERG)在心肌细胞中传导快速外向复极化钾电流(IKr)。由于 hERG 与许多主要靶标的结构-活性关系经常重叠,因此药物样分子对该通道的意外阻断是药物研发的一个关键挑战。基于我们之前的工作,以及最近 hERG 的冷冻电镜结构,我们着手更好地理解在生物动力学理论背景下,混杂阻断剂与 hERG 结合的能量和结构基础。我们提出了一个两步阻断剂结合过程,包括:初始捕获步骤:单个完全溶剂化的阻断剂副本扩散到由细胞内环核苷酸结合同源结构域(CNBHD)排列的大腔中。占据这个腔是离子电流中断的必要但不充分条件。IKr 中断步骤:捕获的阻断剂沿着通道轴的易位,使得:头部基团,由准棒状部分组成,投射到开放的孔中,伴随着结合界面的部分去溶剂化。一个尾部部分沿着 S6 螺旋和靠近孔细胞内入口的近端 C 接头螺旋之间的扭结堆积,同样伴随着结合界面的相互去溶剂化(请注意,结合势垒主要由总头部+尾部基团去溶剂化成本组成)。含有高度平面部分的阻断剂,该部分投射到封闭通道内的一个假定狭窄区域内,在通道关闭时被捕获,也在该区域之前终止的阻断剂被捕获。单个捕获的阻断剂副本可能在离开 CNBHD 腔之前多次与孔结合和解离。最后,我们强调当前 hERG 安全指数(SI)的可能缺陷,并提出了一种替代的体内相关策略,考虑到:获益/风险。基于对未患病人心室肌细胞动作电位(AP)模拟的预测致心律失常性 hERG 占有率分数。由于潜在疾病导致安全阈值改变。由于患者间药代动力学(PK)变异性、药物相互作用、过量用药以及用于标签外适应症(其中 hERG 安全参数可能与其标签内对应物不同)而导致暴露水平向预测的心律失常极限升高的风险。