Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, 9201 University City Blvd, Charlotte, NC, 28223, USA.
BMC Med Genomics. 2020 Nov 10;13(1):170. doi: 10.1186/s12920-020-00818-6.
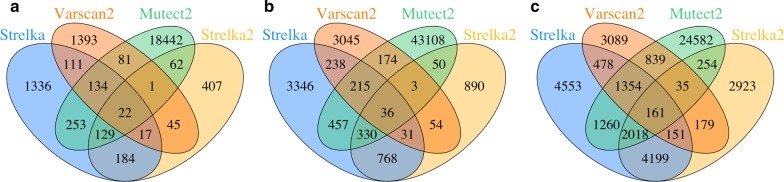
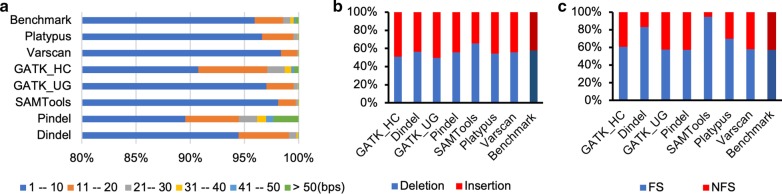
Insertion and deletion (indel) is one of the major variation types in human genomes. Accurate annotation of indels is of paramount importance in genetic variation analysis and investigation of their roles in human diseases. Previous studies revealed a high number of false positives from existing indel calling methods, which limits downstream analyses of the effects of indels on both healthy and disease genomes. In this study, we evaluated seven commonly used general indel calling programs for germline indels and four somatic indel calling programs through comparative analysis to investigate their common features and differences and to explore ways to improve indel annotation accuracy.
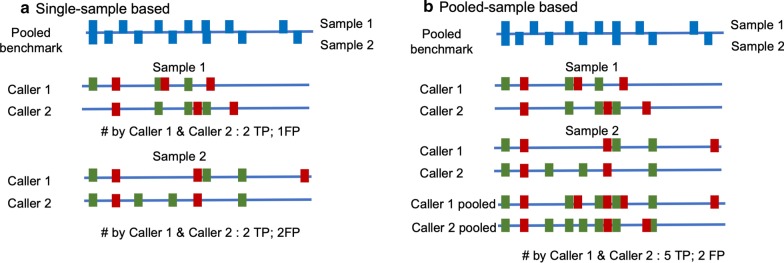
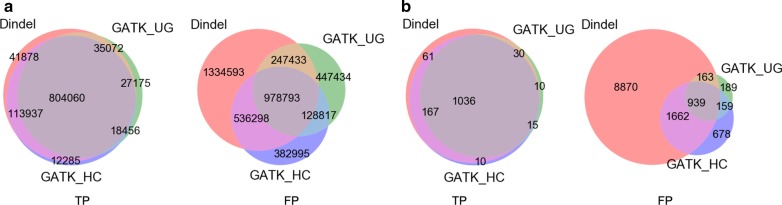
In our comparative analysis, we adopted a more stringent evaluation approach by considering both the indel positions and the indel types (insertion or deletion sequences) between the samples and the reference set. In addition, we applied an efficient way to use a benchmark for improved performance comparisons for the general indel calling programs RESULTS: We found that germline indels in healthy genomes derived by combining several indel calling tools could help remove a large number of false positive indels from individual programs without compromising the number of true positives. The performance comparisons of somatic indel calling programs are more complicated due to the lack of a reliable and comprehensive benchmark. Nevertheless our results revealed large variations among the programs and among cancer types.
While more accurate indel calling programs are needed, we found that the performance for germline indel annotations can be improved by combining the results from several programs. In addition, well-designed benchmarks for both germline and somatic indels are key in program development and evaluations.
插入和缺失(indel)是人类基因组中的主要变异类型之一。准确注释 indel 在遗传变异分析和研究其在人类疾病中的作用方面至关重要。先前的研究表明,现有 indel 调用方法存在大量的假阳性,这限制了 indel 对健康和疾病基因组影响的下游分析。在这项研究中,我们通过比较分析评估了七种常用的种系 indel 调用程序和四种体细胞 indel 调用程序,以研究它们的共同特征和差异,并探讨提高 indel 注释准确性的方法。
在我们的比较分析中,我们采用了更严格的评估方法,同时考虑了样本和参考集之间的 indel 位置和 indel 类型(插入或缺失序列)。此外,我们采用了一种有效的方法来使用基准来改进通用 indel 调用程序的性能比较。
我们发现,通过组合几种 indel 调用工具获得的健康基因组中的种系 indel 可以帮助从单个程序中去除大量假阳性 indel,而不会影响真阳性的数量。由于缺乏可靠和全面的基准,体细胞 indel 调用程序的性能比较更加复杂。然而,我们的结果揭示了程序之间和癌症类型之间的巨大差异。
虽然需要更准确的 indel 调用程序,但我们发现通过组合多个程序的结果可以提高种系 indel 注释的性能。此外,种系和体细胞 indel 的精心设计的基准是程序开发和评估的关键。