National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, 430070, Wuhan, China.
Hubei Key Laboratory of Agricultural Bioinformatics, Huazhong Agricultural University, 430070, Wuhan, China.
Commun Biol. 2020 Nov 13;3(1):675. doi: 10.1038/s42003-020-01403-4.
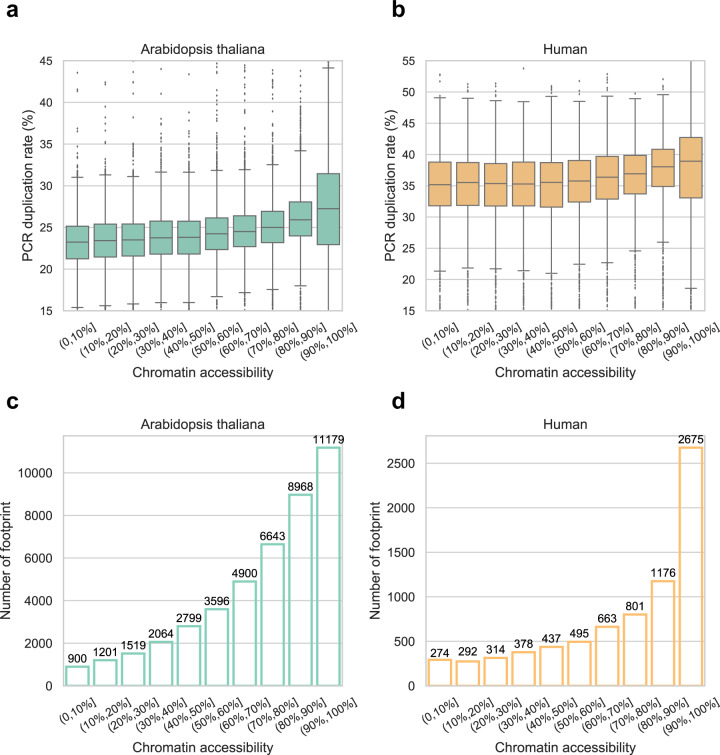
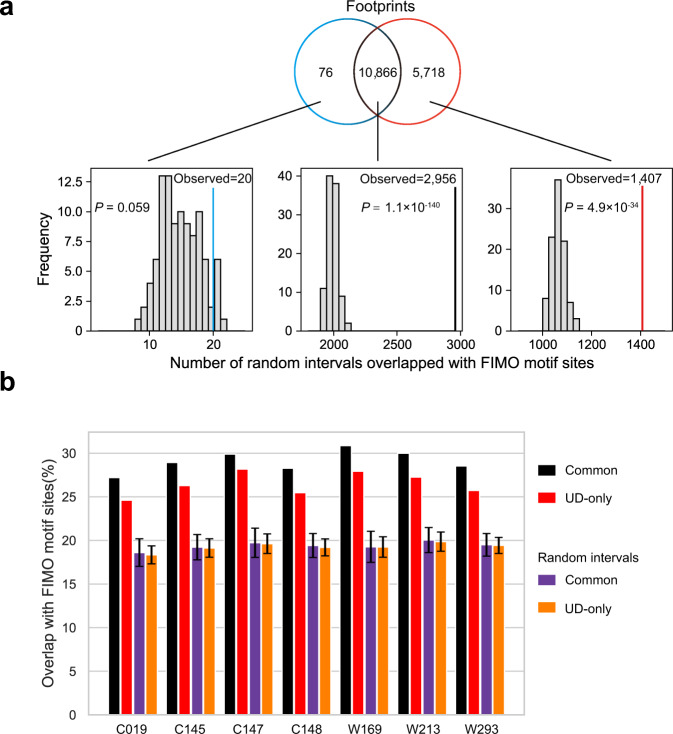
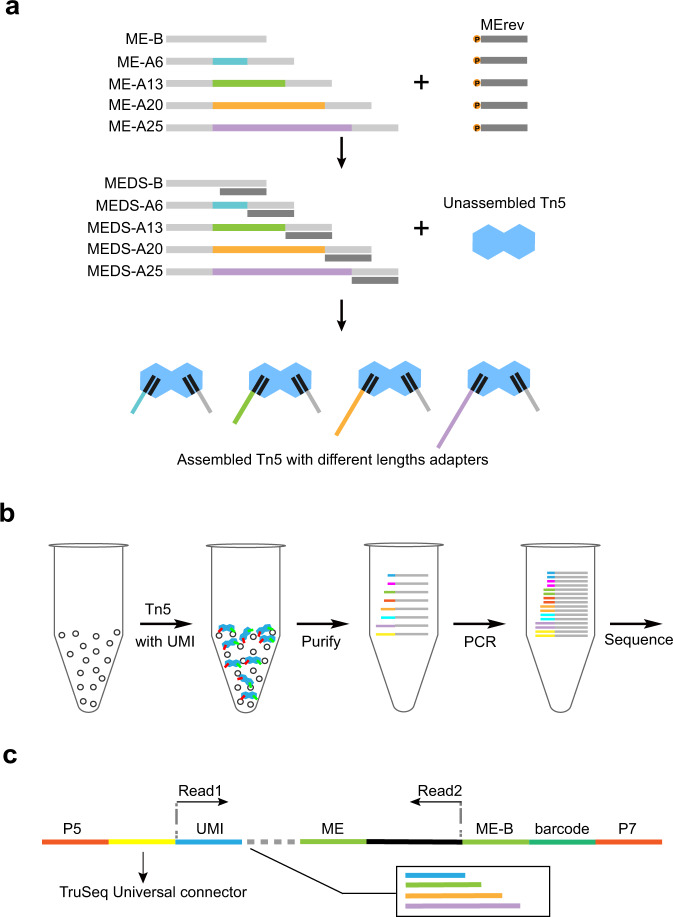
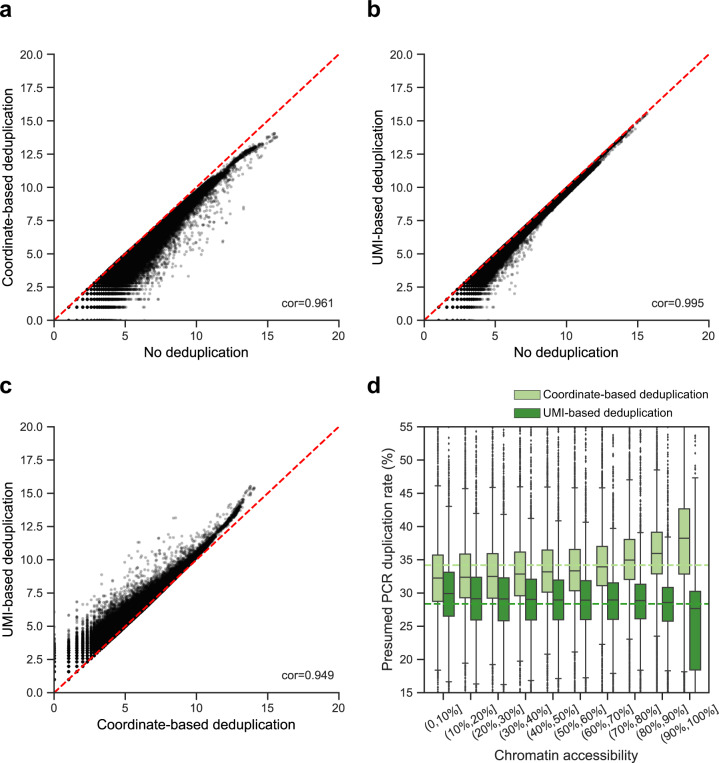
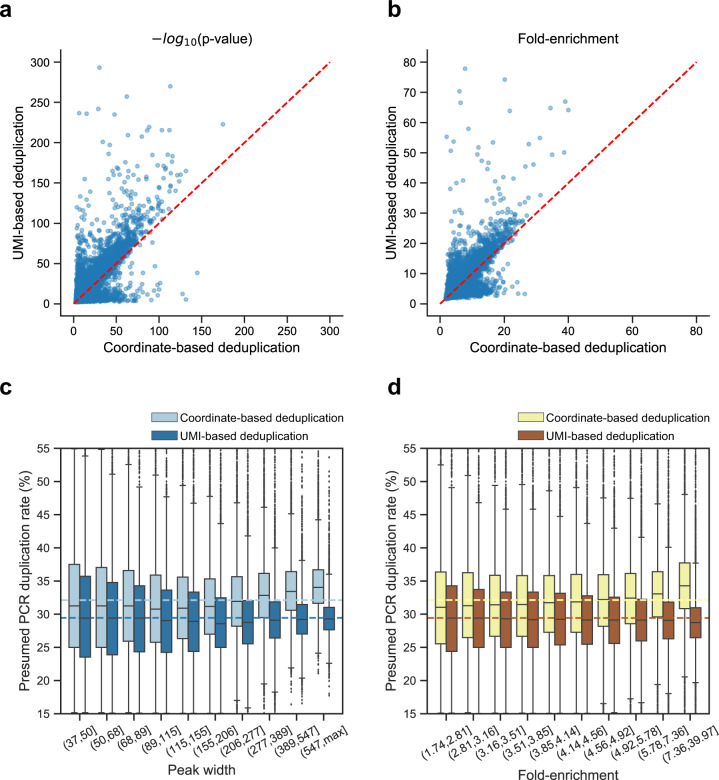
ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) provides an efficient way to analyze nucleosome-free regions and has been applied widely to identify transcription factor footprints. Both applications rely on the accurate quantification of insertion events of the hyperactive transposase Tn5. However, due to the presence of the PCR amplification, it is impossible to accurately distinguish independently generated identical Tn5 insertion events from PCR duplicates using the standard ATAC-seq technique. Removing PCR duplicates based on mapping coordinates introduces increasing bias towards highly accessible chromatin regions. To overcome this limitation, we establish a UMI-ATAC-seq technique by incorporating unique molecular identifiers (UMIs) into standard ATAC-seq procedures. UMI-ATAC-seq can rescue about 20% of reads that are mistaken as PCR duplicates in standard ATAC-seq in our study. We demonstrate that UMI-ATAC-seq could more accurately quantify chromatin accessibility and significantly improve the sensitivity of identifying transcription factor footprints. An analytic pipeline is developed to facilitate the application of UMI-ATAC-seq, and it is available at https://github.com/tzhu-bio/UMI-ATAC-seq .
ATAC-seq(高通量测序的转座酶可及染色质分析)提供了一种分析无核小体区域的有效方法,并已广泛应用于鉴定转录因子足迹。这两种应用都依赖于超活性转座酶 Tn5 插入事件的准确定量。然而,由于存在 PCR 扩增,使用标准 ATAC-seq 技术不可能从 PCR 重复中准确区分独立生成的相同 Tn5 插入事件。基于映射坐标去除 PCR 重复会导致对高度可及染色质区域的偏向增加。为了克服这一限制,我们通过将独特分子标识符 (UMI) 纳入标准 ATAC-seq 程序,建立了 UMI-ATAC-seq 技术。在我们的研究中,UMI-ATAC-seq 可以挽救标准 ATAC-seq 中约 20%被误认为是 PCR 重复的读取。我们证明 UMI-ATAC-seq 可以更准确地定量染色质可及性,并显著提高鉴定转录因子足迹的灵敏度。开发了一个分析管道来促进 UMI-ATAC-seq 的应用,可在 https://github.com/tzhu-bio/UMI-ATAC-seq 上获得。