Milocco Francesca, de Vries Folkert, Bartels Imke M A, Havenith Remco W A, Cirera Jordi, Demeshko Serhiy, Meyer Franc, Otten Edwin
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands.
Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands.
J Am Chem Soc. 2020 Nov 25;142(47):20170-20181. doi: 10.1021/jacs.0c10010. Epub 2020 Nov 16.
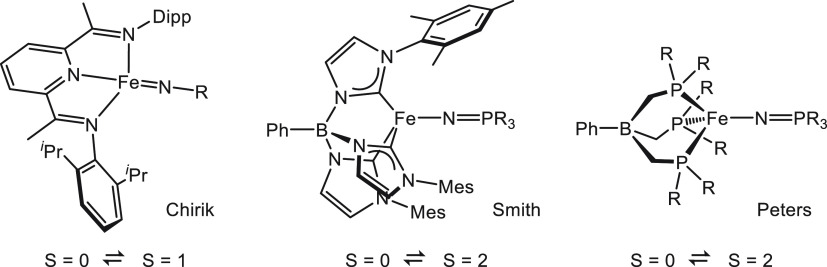
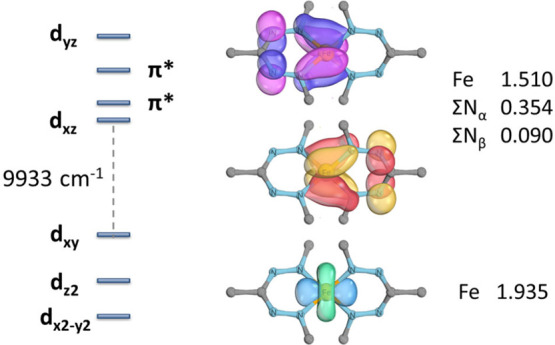
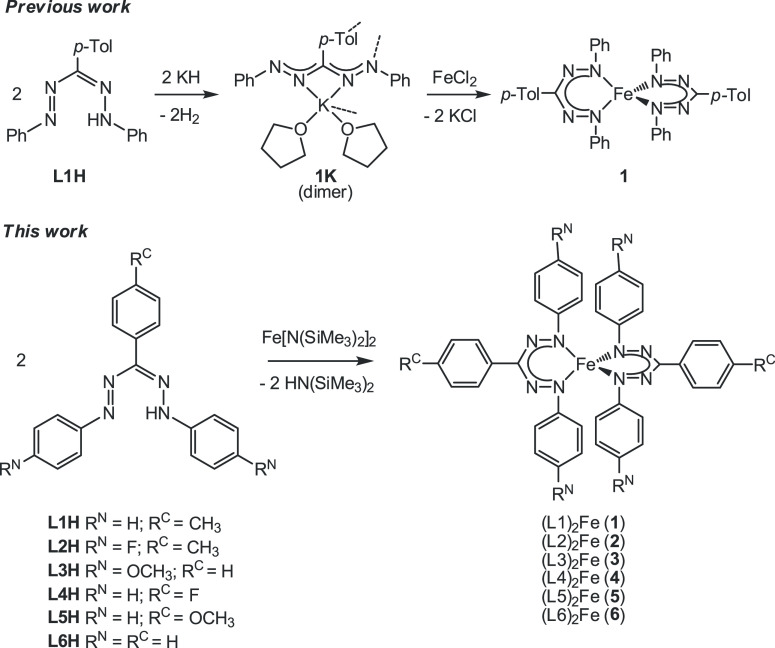
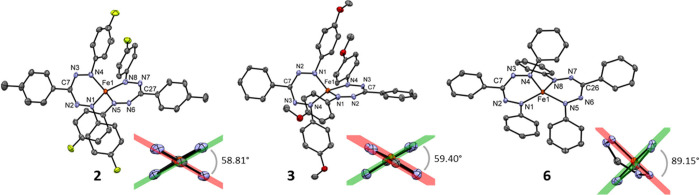
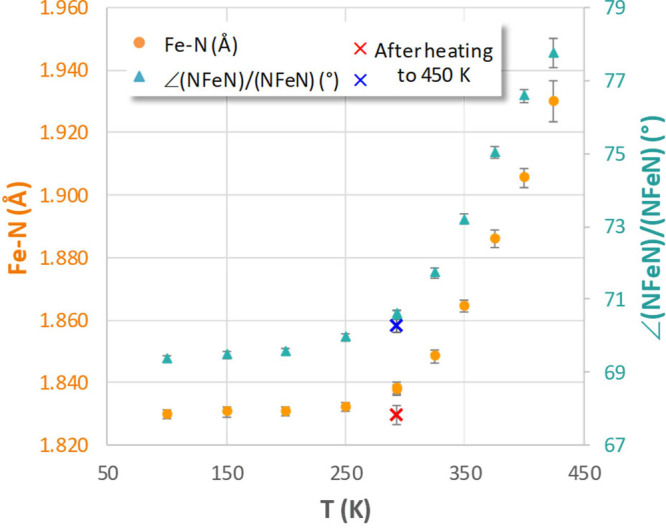
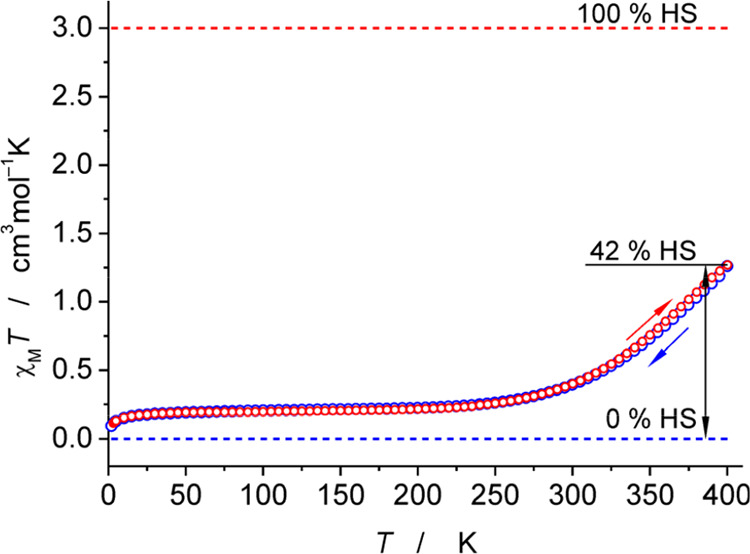
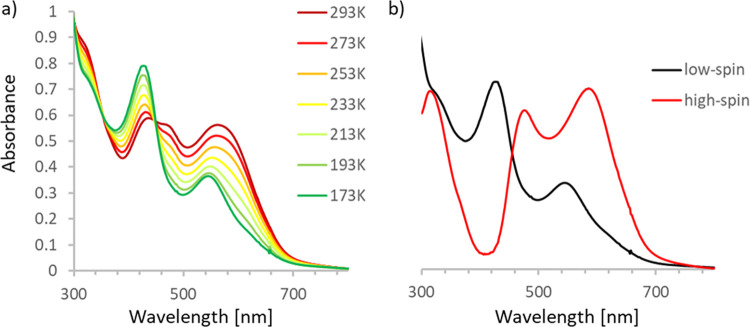
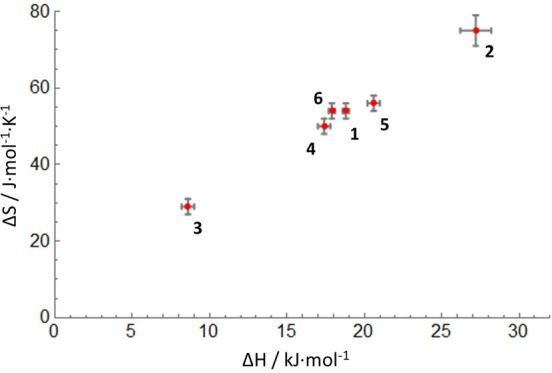
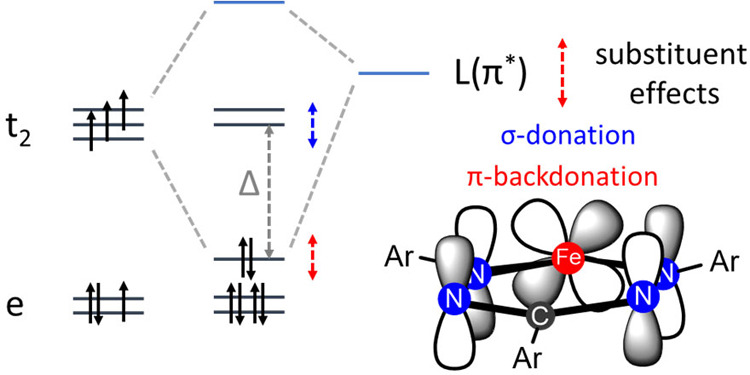
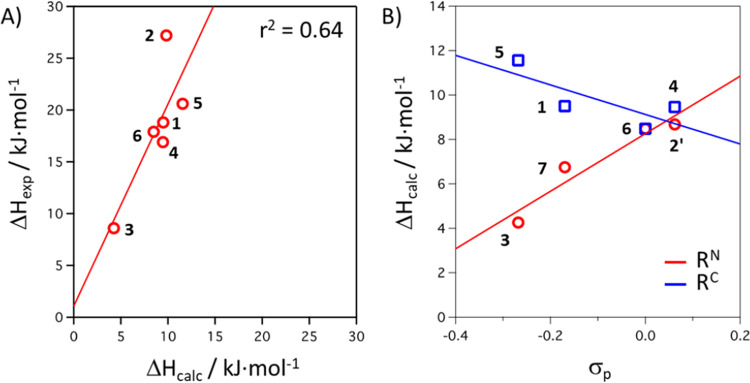
The transition between spin states in d-block metal complexes has important ramifications for their structure and reactivity, with applications ranging from information storage materials to understanding catalytic activity of metalloenzymes. Tuning the ligand field (Δ) by steric and/or electronic effects has provided spin-crossover compounds for several transition metals in the periodic table, but this has mostly been limited to coordinatively saturated metal centers in octahedral ligand environments. Spin-crossover complexes with low coordination numbers are much rarer. Here we report a series of four-coordinate, (pseudo)tetrahedral Fe(II) complexes with formazanate ligands and demonstrate how electronic substituent effects can be used to modulate the thermally induced transition between = 0 and = 2 spin states in solution. All six compounds undergo spin-crossover in solution with above room temperature (300-368 K). While structural analysis by X-ray crystallography shows that the majority of these compounds are low-spin in the solid state (and remain unchanged upon heating), we find that packing effects can override this preference and give rise to either rigorously high-spin () or gradual spin-crossover behavior () also in the solid state. Density functional theory calculations are used to delineate the empirical trends in solution spin-crossover thermodynamics. In all cases, the stabilization of the low-spin state is due to the π-acceptor properties of the formazanate ligand, resulting in an "inverted" ligand field, with an approximate "two-over-three" splitting of the d-orbitals and a high degree of metal-ligand covalency due to metal → ligand π-backdonation. The computational data indicate that the electronic nature of the -substituent has a different influence depending on whether it is present at the C-Ar or N-Ar rings, which is ascribed to the opposing effect on metal-ligand σ- and π-bonding.
d 族金属配合物中自旋态之间的转变对其结构和反应性具有重要影响,其应用范围涵盖从信息存储材料到理解金属酶的催化活性等领域。通过空间和/或电子效应调节配体场(Δ)已为元素周期表中的几种过渡金属提供了自旋交叉化合物,但这大多局限于八面体配体环境中配位饱和的金属中心。低配位数的自旋交叉配合物则更为罕见。在此,我们报道了一系列具有甲脒配体的四配位(伪)四面体 Fe(II) 配合物,并展示了如何利用电子取代基效应来调节溶液中 S = 0 和 S = 2 自旋态之间的热诱导转变。所有六种化合物在溶液中均在室温以上(300 - 368 K)发生自旋交叉。虽然 X 射线晶体学的结构分析表明这些化合物中的大多数在固态时为低自旋态(加热后保持不变),但我们发现堆积效应可以克服这种偏好,并且在固态时也会产生严格的高自旋态(S = 2)或逐渐的自旋交叉行为(S = 1)。密度泛函理论计算用于描绘溶液自旋交叉热力学中的经验趋势。在所有情况下,低自旋态的稳定是由于甲脒配体的 π 受体性质,导致“反转”的配体场,d 轨道近似“二比三”分裂,并且由于金属→配体 π 反馈键合而具有高度的金属 - 配体共价性。计算数据表明,-取代基的电子性质根据其存在于 C - Ar 环还是 N - Ar 环而具有不同的影响,这归因于对金属 - 配体 σ 键和 π 键的相反作用。