Bandoy Dj Darwin
Department of Veterinary Paraclinical Sciences, University of the Philippines Los Baños, Los Baños, Laguna, 4031, Philippines.
F1000Res. 2019 Jan 9;8:33. doi: 10.12688/f1000research.17620.3. eCollection 2019.
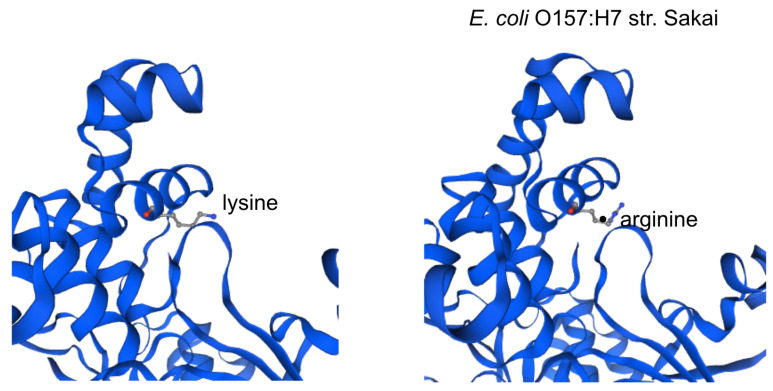
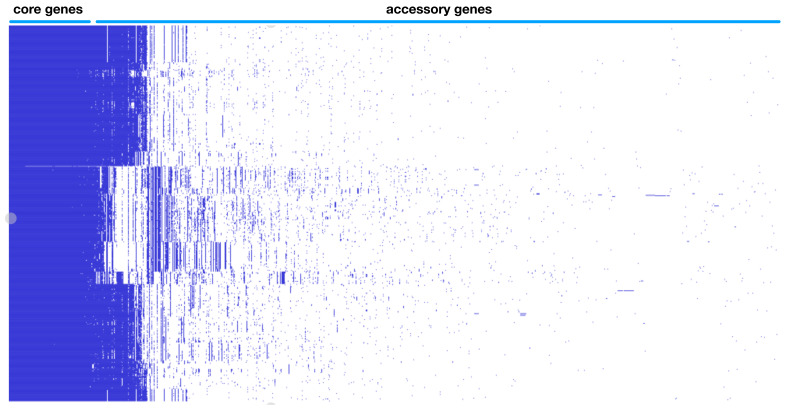
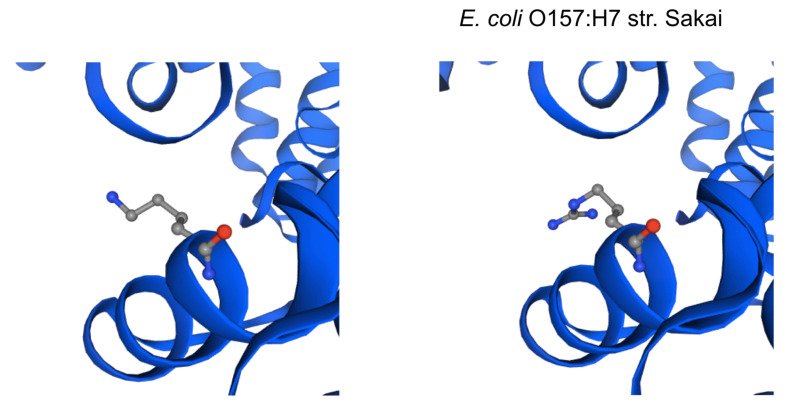
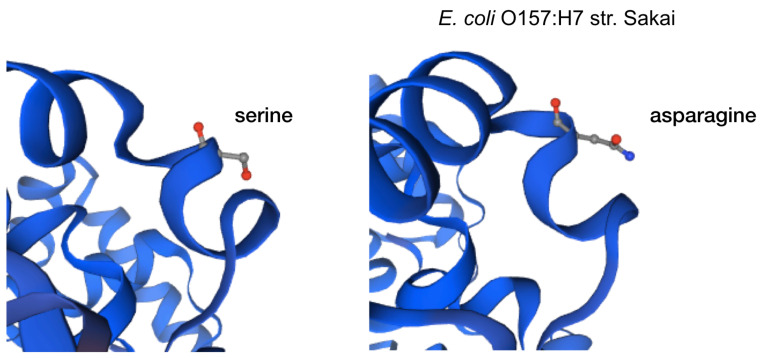
Enterohemorrhagic continues to be a significant public health risk. With the onset of next generation sequencing, whole genome sequences require a new paradigm of analysis relevant for epidemiology and drug discovery. A large-scale bacterial population genomic analysis was applied to 702 isolates of serotypes associated with EHEC resulting in five pangenome clusters. Serotype incongruence with pangenome types suggests recombination clusters. Core genome analysis was performed to determine the population wide distribution of sdiA as potential drug target. Protein modelling revealed nonsynonymous variants are notably absent in the ligand binding site for quorum sensing, indicating that population wide conservation of the sdiA ligand site can be targeted for potential prophylactic purposes. Applying pathotype-wide pangenomics as a guide for determining evolution of pharmacophore sites is a potential approach in drug discovery.
肠出血性大肠杆菌仍然是一个重大的公共卫生风险。随着下一代测序技术的出现,全基因组序列需要一种适用于流行病学和药物发现的新分析范式。对702株与肠出血性大肠杆菌相关血清型的分离株进行了大规模细菌群体基因组分析,结果产生了五个泛基因组簇。血清型与泛基因组类型不一致表明存在重组簇。进行核心基因组分析以确定群体中作为潜在药物靶点的sdiA的分布。蛋白质建模显示群体感应的配体结合位点中明显不存在非同义变体,这表明sdiA配体位点在群体中的保守性可作为潜在预防目的的靶点。将致病型泛基因组学作为确定药效团位点进化的指南是药物发现中的一种潜在方法。