Bandoy D J Darwin R, Weimer Bart C
School of Veterinary Medicine, Population Health and Reproduction, 100K Pathogen Genome Project, University of California Davis, Davis, CA, 95616, USA.
Department of Veterinary Paraclinical Sciences, College of Veterinary Medicine, University of the Philippines Los Baños, 4031, Los Baños, Laguna, Philippines.
Sci Rep. 2021 Apr 1;11(1):7380. doi: 10.1038/s41598-021-86265-4.
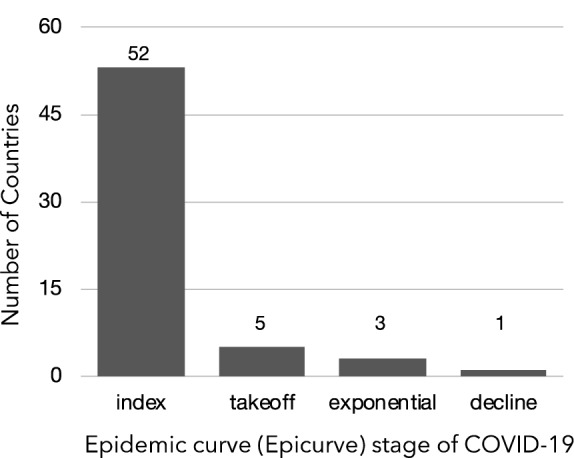
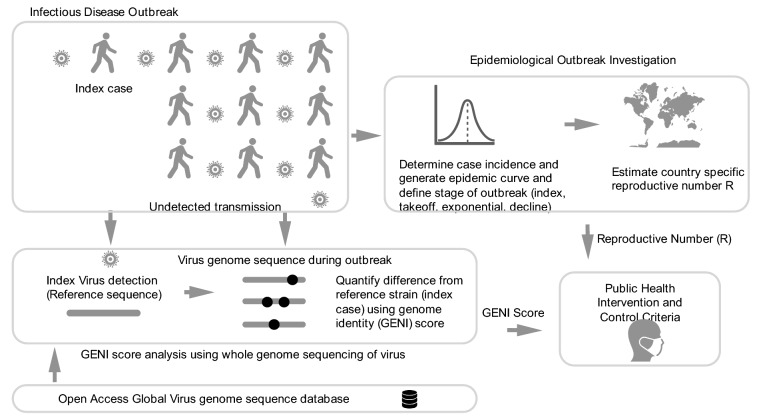
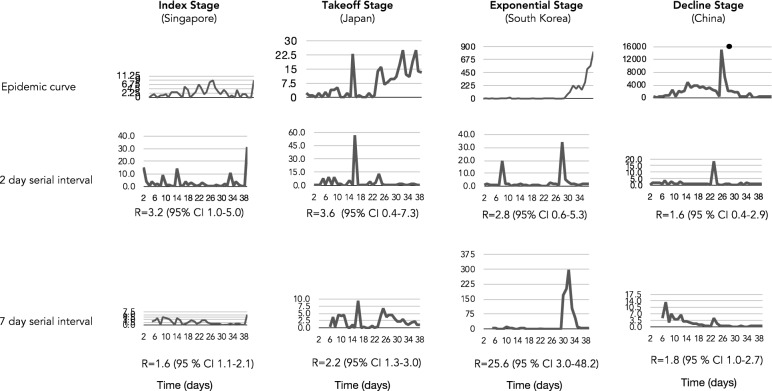
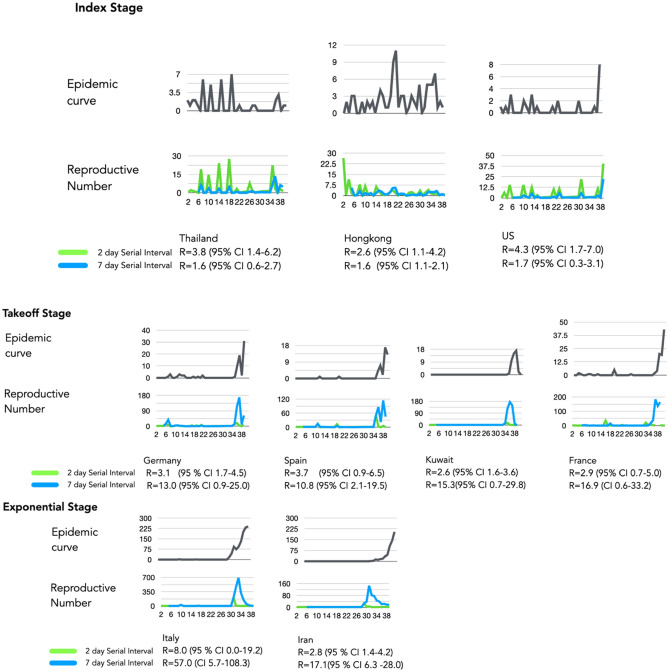
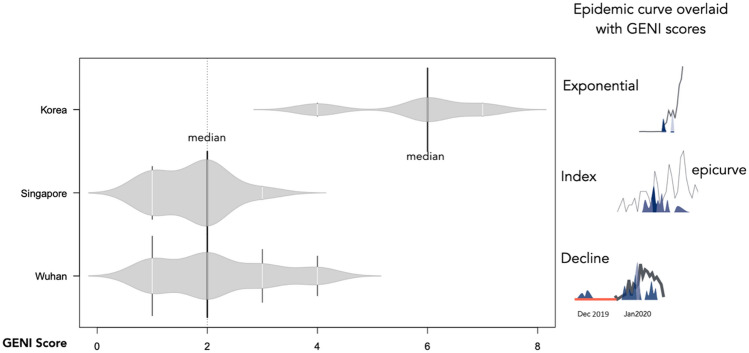
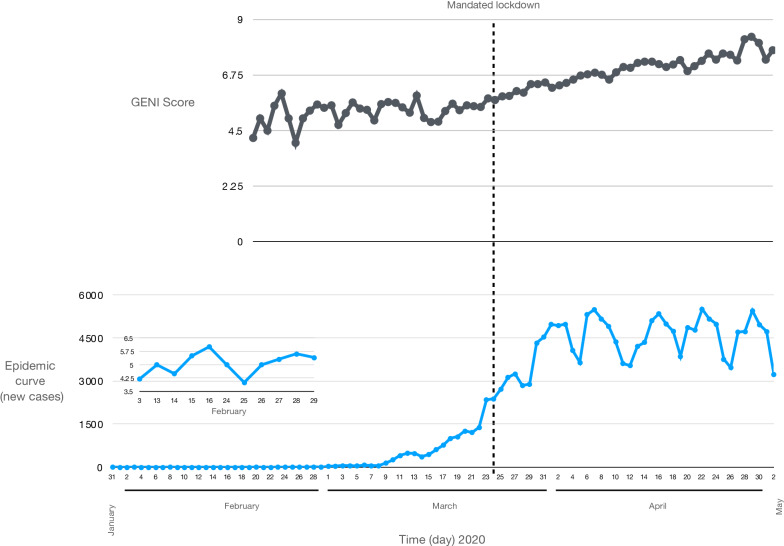
The spread of SARS-CoV-2 created a pandemic crisis with > 150,000 cumulative cases in > 65 countries within a few months. The reproductive number (R) is a metric to estimate the transmission of a pathogen during an outbreak. Preliminary published estimates were based on the initial outbreak in China. Whole genome sequences (WGS) analysis found mutational variations in the viral genome; however, previous comparisons failed to show a direct relationship between viral genome diversity, transmission, and the epidemic severity. COVID-19 incidences from different countries were modeled over the epidemic curve. Estimates of the instantaneous R (Wallinga and Teunis method) with a short and standard serial interval were done. WGS were used to determine the populations genomic variation and that underpinned creation of the pathogen genome identity (GENI) score, which was merged with the outbreak curve in four distinct phases. Inference of transmission time was based on a mutation rate of 2 mutations/month. R estimates revealed differences in the transmission and variable infection dynamics between and within outbreak progression for each country examined. Outside China, our R estimates observed propagating dynamics indicating that other countries were poised to move to the takeoff and exponential stages. Population density and local temperatures had no clear relationship to the outbreak progression. Integration of incidence data with the GENI score directly predicted increases in cases as the genome variation increased that led to new variants. Integrating the outbreak curve, dynamic R, and SNP variation found a direct association between increasing cases and transmission genome evolution. By defining the epidemic curve into four stages and integrating the instantaneous country-specific R with the GENI score, we directly connected changes in individual outbreaks based on changes in the virus genome via SNPs. This resulted in the ability to forecast potential increases in cases as well as mutations that may defeat PCR screening and the infection process. By using instantaneous R estimations and WGS, outbreak dynamics were defined to be linked to viral mutations, indicating that WGS, as a surveillance tool, is required to predict shifts in each outbreak that will provide actionable decision making information. Integrating epidemiology with genome sequencing and modeling allows for evidence-based disease outbreak tracking with predictive therapeutically valuable insights in near real time.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的传播在几个月内造成了一场大流行危机,65多个国家累计病例超过15万例。繁殖数(R)是评估病原体在疫情爆发期间传播情况的一个指标。已发表的初步估计是基于中国的首次疫情爆发。全基因组序列(WGS)分析发现病毒基因组存在突变变异;然而,先前的比较未能显示病毒基因组多样性、传播与疫情严重程度之间的直接关系。对来自不同国家的2019冠状病毒病发病率进行了疫情曲线建模。采用短和标准潜伏期的瞬时R(瓦林加和特尼斯方法)进行了估计。利用全基因组测序来确定群体基因组变异,并以此为基础创建病原体基因组身份(GENI)评分,该评分与四个不同阶段的疫情曲线相结合。传播时间的推断基于每月2次突变的突变率。R估计值揭示了每个被研究国家在疫情发展过程中不同国家之间以及国家内部传播和可变感染动态的差异。在中国境外,我们对R的估计观察到传播动态,表明其他国家正准备进入爆发期和指数增长期。人口密度和当地温度与疫情发展没有明显关系。将发病率数据与GENI评分相结合,直接预测了随着基因组变异增加导致新变种而出现的病例增加情况。整合疫情曲线、动态R和单核苷酸多态性(SNP)变异发现病例增加与传播基因组进化之间存在直接关联。通过将疫情曲线划分为四个阶段,并将特定国家的瞬时R与GENI评分相结合,我们基于单核苷酸多态性通过病毒基因组的变化直接关联了各个疫情的变化。这使得能够预测病例的潜在增加以及可能使聚合酶链反应(PCR)筛查和感染过程失效的突变。通过使用瞬时R估计值和全基因组测序,确定疫情动态与病毒突变有关,这表明全基因组测序作为一种监测工具,对于预测每次疫情的变化是必要的,而这些变化将提供可采取行动的决策信息。将流行病学与基因组测序和建模相结合,能够以基于证据的方式跟踪疾病爆发,并在近乎实时的情况下获得具有预测性治疗价值的见解。