Division of Medical Microbiology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA.
CosmosID, Inc., Rockville, Maryland, USA.
mBio. 2020 Nov 20;11(6):e01969-20. doi: 10.1128/mBio.01969-20.
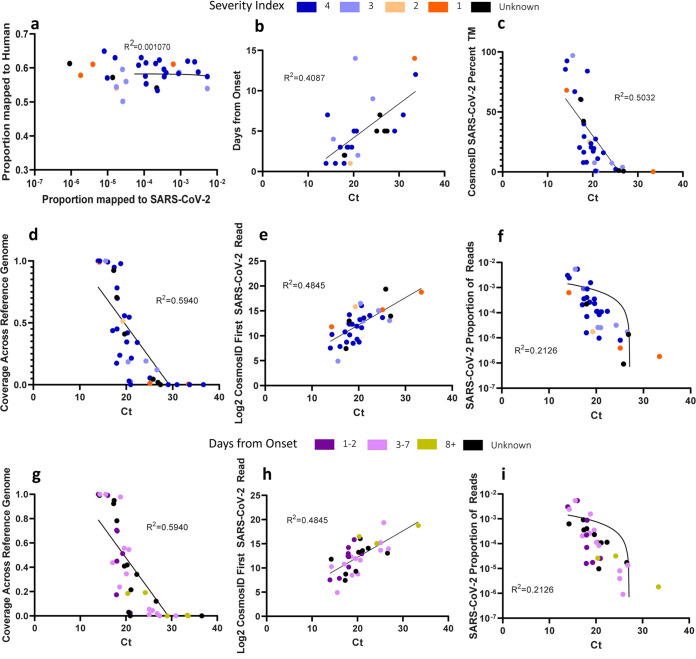
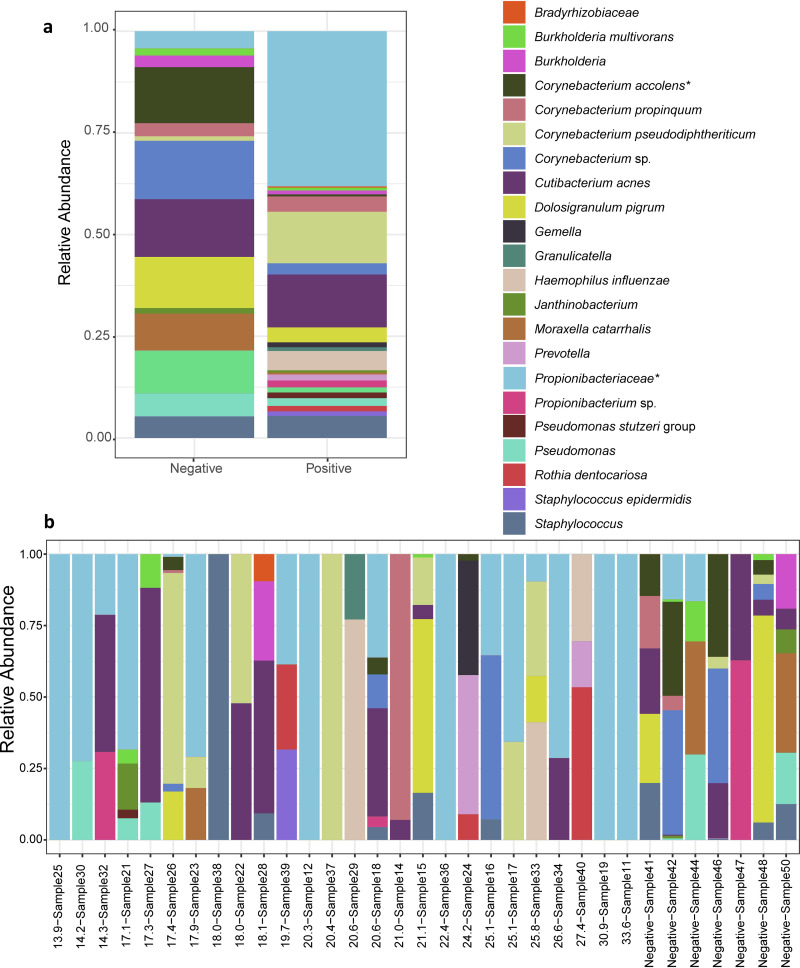
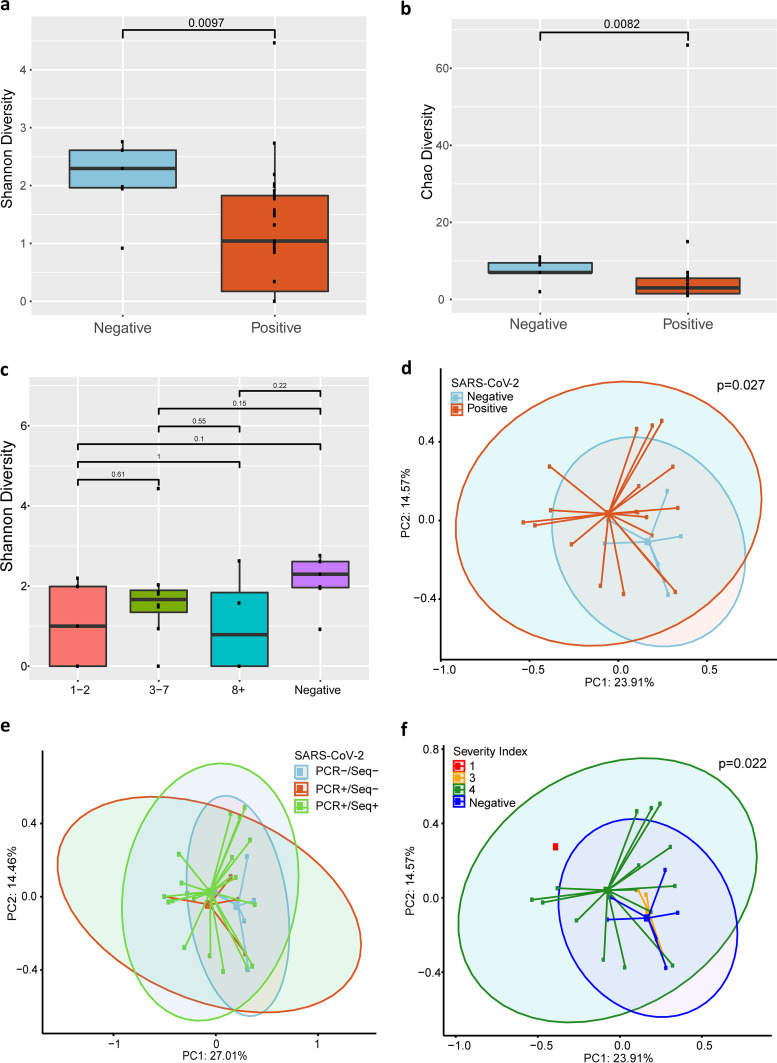
Metagenomic next-generation sequencing (mNGS) offers an agnostic approach for emerging pathogen detection directly from clinical specimens. In contrast to targeted methods, mNGS also provides valuable information on the composition of the microbiome and might uncover coinfections that may associate with disease progression and impact prognosis. To evaluate the use of mNGS for detecting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and/or other infecting pathogens, we applied direct Oxford Nanopore long-read third-generation metatranscriptomic and metagenomic sequencing. Nasopharyngeal (NP) swab specimens from 50 patients under investigation for CoV disease 2019 (COVID-19) were sequenced, and the data were analyzed by the CosmosID bioinformatics platform. Further, we characterized coinfections and the microbiome associated with a four-point severity index. SARS-CoV-2 was identified in 77.5% (31/40) of samples positive by RT-PCR, correlating with lower cycle threshold (Ct) values and fewer days from symptom onset. At the time of sampling, possible bacterial or viral coinfections were detected in 12.5% of SARS-CoV-2-positive specimens. A decrease in microbial diversity was observed among COVID-19-confirmed patients (Shannon diversity index, = 0.0082; Chao richness estimate, = 0.0097; Simpson diversity index, = 0.018), and differences in microbial communities were linked to disease severity ( = 0.022). Furthermore, statistically significant shifts in the microbiome were identified among SARS-CoV-2-positive and -negative patients, in the latter of whom a higher abundance of ( = 0.028) and a reduction in the abundance of ( = 0.025) were observed. Our study corroborates the growing evidence that increased SARS-CoV-2 RNA detection from NP swabs is associated with the early stages rather than the severity of COVID-19. Further, we demonstrate that SARS-CoV-2 causes a significant change in the respiratory microbiome. This work illustrates the utility of mNGS for the detection of SARS-CoV-2, for diagnosing coinfections without viral target enrichment or amplification, and for the analysis of the respiratory microbiome. SARS-CoV-2 has presented a rapidly accelerating global public health crisis. The ability to detect and analyze viral RNA from minimally invasive patient specimens is critical to the public health response. Metagenomic next-generation sequencing (mNGS) offers an opportunity to detect SARS-CoV-2 from nasopharyngeal (NP) swabs. This approach also provides information on the composition of the respiratory microbiome and its relationship to coinfections or the presence of other organisms that may impact SARS-CoV-2 disease progression and prognosis. Here, using direct Oxford Nanopore long-read third-generation metatranscriptomic and metagenomic sequencing of NP swab specimens from 50 patients under investigation for COVID-19, we detected SARS-CoV-2 sequences by applying the CosmosID bioinformatics platform. Further, we characterized coinfections and detected a decrease in the diversity of the microbiomes in these patients. Statistically significant shifts in the microbiome were identified among COVID-19-positive and -negative patients, in the latter of whom a higher abundance of and a reduction in the abundance of were observed. Our study also corroborates the growing evidence that increased SARS-CoV-2 RNA detection from NP swabs is associated with the early stages of disease rather than with severity of disease. This work illustrates the utility of mNGS for the detection and analysis of SARS-CoV-2 from NP swabs without viral target enrichment or amplification and for the analysis of the respiratory microbiome.
宏基因组下一代测序 (mNGS) 为直接从临床标本中检测新兴病原体提供了一种无偏倚的方法。与靶向方法相比,mNGS 还提供了微生物组组成的有价值信息,并可能揭示可能与疾病进展和预后相关的合并感染。为了评估直接 Oxford Nanopore 长读长第三代宏转录组和宏基因组测序用于检测严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 和/或其他感染病原体的用途,我们对 50 名接受 2019 年冠状病毒病 (COVID-19) 调查的患者的鼻咽 (NP) 拭子标本进行了测序,并通过 CosmosID 生物信息学平台对数据进行了分析。此外,我们还对与四点严重指数相关的合并感染和微生物组进行了特征描述。通过 RT-PCR 检测到 77.5% (31/40) 的 SARS-CoV-2 阳性样本,这与较低的循环阈值 (Ct) 值和发病后天数较少相关。在采样时,在 SARS-CoV-2 阳性样本中检测到可能的细菌或病毒合并感染 12.5%。COVID-19 确诊患者的微生物多样性下降 (Shannon 多样性指数, = 0.0082;Chao 丰富度估计, = 0.0097;Simpson 多样性指数, = 0.018),微生物群落的差异与疾病严重程度相关 ( = 0.022)。此外,在 SARS-CoV-2 阳性和阴性患者之间鉴定到微生物组的统计学显著变化,在后一组中观察到更高的丰度 ( = 0.028) 和丰度降低 ( = 0.025)。我们的研究证实了越来越多的证据,即从 NP 拭子中检测到的 SARS-CoV-2 RNA 增加与 COVID-19 的早期阶段而不是严重程度相关。此外,我们证明 SARS-CoV-2 导致呼吸道微生物组发生显著变化。这项工作说明了 mNGS 用于检测 SARS-CoV-2 的实用性,用于在没有病毒靶标富集或扩增的情况下诊断合并感染,以及用于分析呼吸道微生物组。SARS-CoV-2 引发了迅速加速的全球公共卫生危机。从微创患者标本中检测和分析病毒 RNA 的能力对公共卫生应对至关重要。宏基因组下一代测序 (mNGS) 提供了从鼻咽 (NP) 拭子中检测 SARS-CoV-2 的机会。这种方法还提供了有关呼吸道微生物组组成及其与合并感染或存在其他可能影响 SARS-CoV-2 疾病进展和预后的生物体的关系的信息。在这里,我们使用直接 Oxford Nanopore 长读长第三代宏转录组和宏基因组测序对 50 名接受 COVID-19 调查的患者的 NP 拭子标本进行了测序,并通过应用 CosmosID 生物信息学平台检测到了 SARS-CoV-2 序列。此外,我们还对合并感染进行了特征描述,并检测到这些患者的微生物组多样性下降。在 COVID-19 阳性和阴性患者之间鉴定到了微生物组的统计学显著变化,在后一组中观察到更高的丰度 ( = 0.028) 和丰度降低 ( = 0.025)。我们的研究还证实了越来越多的证据,即从 NP 拭子中检测到的 SARS-CoV-2 RNA 增加与疾病的早期阶段而不是严重程度相关。这项工作说明了 mNGS 用于检测和分析 NP 拭子中 SARS-CoV-2 的实用性,而无需病毒靶标富集或扩增,也用于分析呼吸道微生物组。