Piras Rossella, Iatropoulos Paraskevas, Bresin Elena, Todeschini Marta, Gastoldi Sara, Valoti Elisabetta, Alberti Marta, Mele Caterina, Galbusera Miriam, Cuccarolo Paola, Benigni Ariela, Remuzzi Giuseppe, Noris Marina
Clinical Research Center for Rare Diseases 'Aldo e Cele Daccò,' Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Bergamo, Italy.
Front Med (Lausanne). 2020 Nov 3;7:579418. doi: 10.3389/fmed.2020.579418. eCollection 2020.
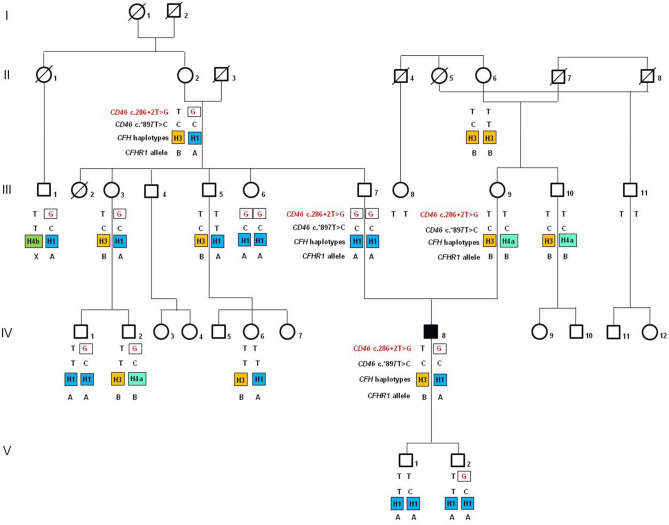
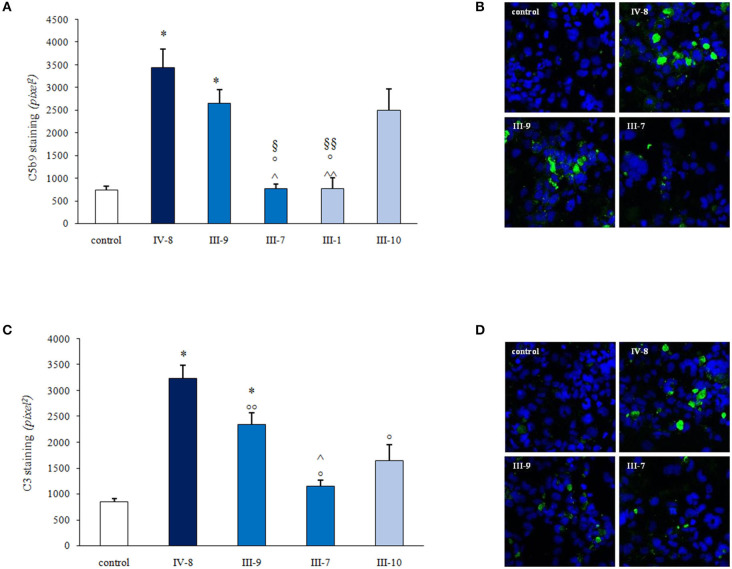
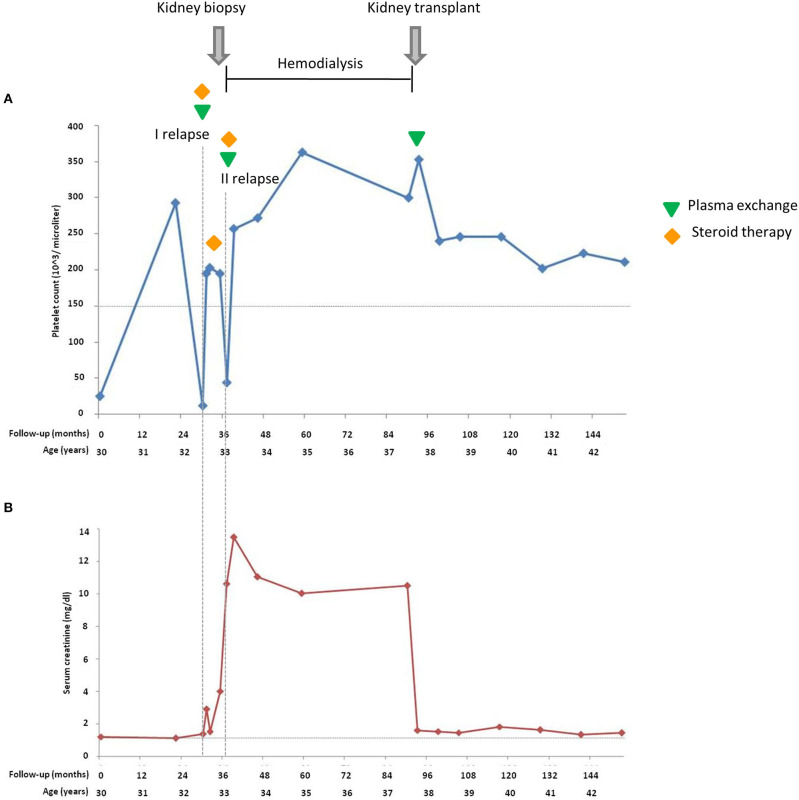
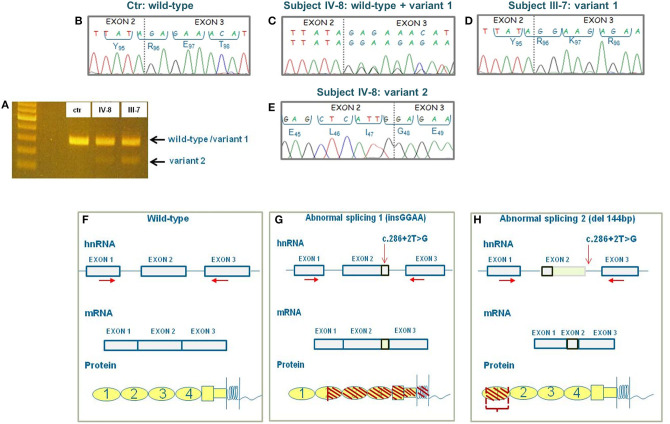
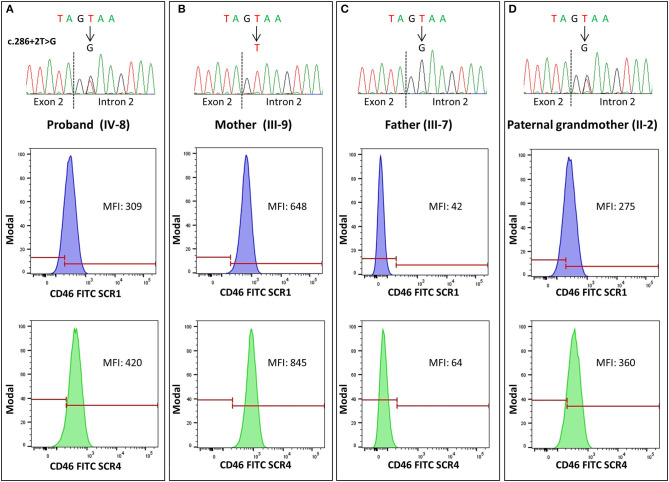
Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disease characterized by microangiopathic hemolysis, thrombocytopenia, and renal impairment and is associated with dysregulation of the alternative complement pathway on the microvascular endothelium. Outcomes have improved greatly with pharmacologic complement C5 blockade. Abnormalities in complement genes (, and ), genomic rearrangements, and anti-FH antibodies have been reported in 40-60% of cases. The penetrance of aHUS is incomplete in carriers of complement gene abnormalities; and multiple hits, including the and risk haplotypes and the risk allele, as well as environmental factors, contribute to disease development. Here, we investigated the determinants of penetrance of aHUS associated with genetic abnormalities. We studied 485 aHUS patients and found rare variants (RVs) in about 10%. The c.286+2T>G RV was the most prevalent (13/485) and was associated with <30% penetrance. We conducted an in-depth study of a large pedigree including a proband who is heterozygous for the c.286+2T>G RV who experienced a severe form of aHUS and developed end-stage renal failure. The father and paternal uncle with the same variant in homozygosity and six heterozygous relatives are unaffected. Flow cytometry analysis showed about 50% reduction of CD46 expression on blood mononuclear cells from the heterozygous proband and over 90% reduction in cells from the proband's unaffected homozygous father and aunt. Further genetic studies did not reveal RVs in known aHUS-associated genes or common genetic modifiers that segregated with the disease. Importantly, a specific test showed excessive complement deposition on endothelial cells exposed to sera from the proband, and also from his mother and maternal uncle, who do not carry the c.286+2T>G RV, indicating that they share a circulating defect that results in complement dysregulation on the endothelium. These results highlight the complexity of the genetics of aHUS and indicate that deficiency may not be enough to induce aHUS. We hypothesize that the proband inherited from his mother a genetic abnormality in a complement circulating factor that has not been identified yet, which synergized with the RV in predisposing him to the aHUS phenotype.
非典型溶血性尿毒症综合征(aHUS)是一种极为罕见的疾病,其特征为微血管病性溶血、血小板减少和肾功能损害,与微血管内皮细胞上替代补体途径的失调有关。药物性补体C5阻断使治疗结果有了很大改善。在40%-60%的病例中报告了补体基因( 、 和 )异常、基因组重排以及抗因子H抗体。补体基因异常携带者中aHUS的外显率不完全;多个因素,包括 和 风险单倍型以及 风险等位基因,还有环境因素,都对疾病发展有影响。在此,我们研究了与 基因异常相关的aHUS外显率的决定因素。我们研究了485例aHUS患者,发现约10%存在罕见变异(RVs)。c.286+2T>G RV最为常见(485例中有13例),且外显率<30%。我们对一个大家系进行了深入研究,该家系的先证者为c.286+2T>G RV杂合子,经历了严重形式的aHUS并发展为终末期肾衰竭。父亲和叔祖父为该变异的纯合子,六个杂合亲属未受影响。流式细胞术分析显示,杂合先证者血液单核细胞上CD46表达降低约50%,先证者未受影响的纯合子父亲和姑姑的细胞上CD46表达降低超过90%。进一步的基因研究未在已知的与aHUS相关的基因或与疾病共分离的常见基因修饰因子中发现RVs。重要的是,一项特定检测显示,暴露于先证者血清的内皮细胞上有过量补体沉积,其母亲和舅舅(他们不携带c.286+2T>G RV)的血清也有此现象,这表明他们存在一种共同的循环缺陷,导致内皮细胞上补体失调。这些结果凸显了aHUS遗传学的复杂性,并表明 缺陷可能不足以诱发aHUS。我们推测,先证者从母亲那里继承了一种尚未确定的补体循环因子的基因异常,该异常与RV协同作用,使他易患aHUS表型。