Department of Biostatistics and Computational Biology, University of Rochester Medical Center, 265 Crittenden Blvd., 14642, Rochester, NY, USA.
Department of Biomedical Genetics, University of Rochester Medical Center, 601 Elmwood Ave., 14642, Rochester, NY, USA.
BMC Bioinformatics. 2020 Nov 26;21(1):545. doi: 10.1186/s12859-020-03807-9.
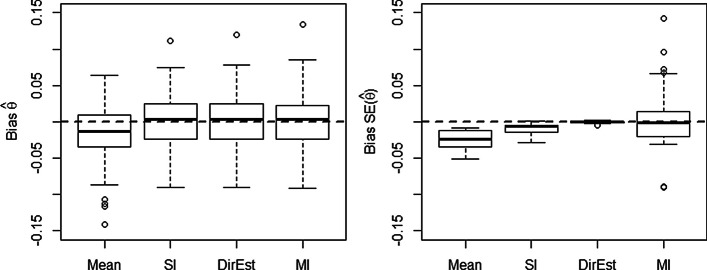
Quantitative real-time PCR (qPCR) is one of the most widely used methods to measure gene expression. An important aspect of qPCR data that has been largely ignored is the presence of non-detects: reactions failing to exceed the quantification threshold and therefore lacking a measurement of expression. While most current software replaces these non-detects with a value representing the limit of detection, this introduces substantial bias in the estimation of both absolute and differential expression. Single imputation procedures, while an improvement on previously used methods, underestimate residual variance, which can lead to anti-conservative inference.
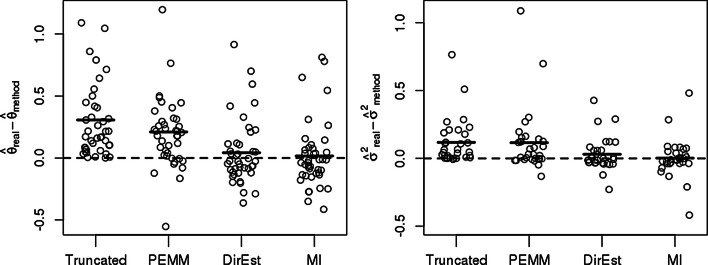
We propose to treat non-detects as non-random missing data, model the missing data mechanism, and use this model to impute missing values or obtain direct estimates of model parameters. To account for the uncertainty inherent in the imputation, we propose a multiple imputation procedure, which provides a set of plausible values for each non-detect. We assess the proposed methods via simulation studies and demonstrate the applicability of these methods to three experimental data sets. We compare our methods to mean imputation, single imputation, and a penalized EM algorithm incorporating non-random missingness (PEMM). The developed methods are implemented in the R/Bioconductor package nondetects.
The statistical methods introduced here reduce discrepancies in gene expression values derived from qPCR experiments in the presence of non-detects, providing increased confidence in downstream analyses.
实时荧光定量 PCR(qPCR)是测量基因表达最广泛使用的方法之一。qPCR 数据的一个重要方面在很大程度上被忽视了,即存在无法检测到的情况:反应未能超过定量阈值,因此缺乏表达的测量。虽然大多数当前的软件用代表检测极限的值替换这些无法检测到的值,但这会对绝对和差异表达的估计引入很大的偏差。单插补程序虽然优于以前使用的方法,但低估了剩余方差,这可能导致反保守的推断。
我们建议将无法检测到的情况视为随机缺失数据,对缺失数据机制进行建模,并使用该模型对缺失值进行插补或直接估计模型参数。为了考虑插补中固有的不确定性,我们提出了一种多重插补程序,为每个无法检测到的值提供一组合理的值。我们通过模拟研究评估了所提出的方法,并展示了这些方法在三个实验数据集上的适用性。我们将我们的方法与均值插补、单插补和纳入非随机缺失的惩罚 EM 算法(PEMM)进行了比较。所开发的方法在 R/Bioconductor 包 nondetects 中实现。
这里介绍的统计方法减少了在存在无法检测到的情况下从 qPCR 实验中得出的基因表达值之间的差异,为下游分析提供了更大的信心。