Bioinformatics Unit, Department of Infectious Diseases, National Institute of Health, 1649-016 Lisbon, Portugal.
Laboratory of Microbiology, Department of Infectious Diseases, National Institute of Health, 1649-016 Lisbon, Portugal.
Microb Genom. 2021 Feb;7(2). doi: 10.1099/mgen.0.000481.
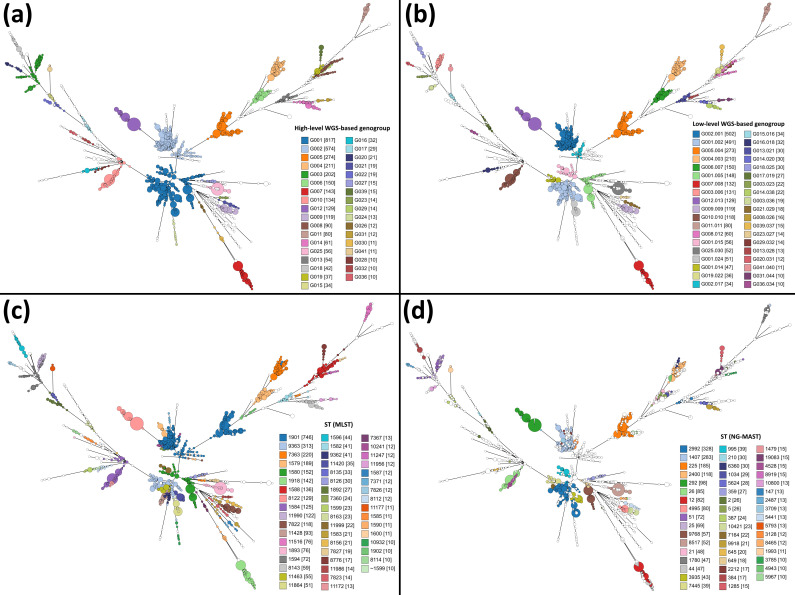
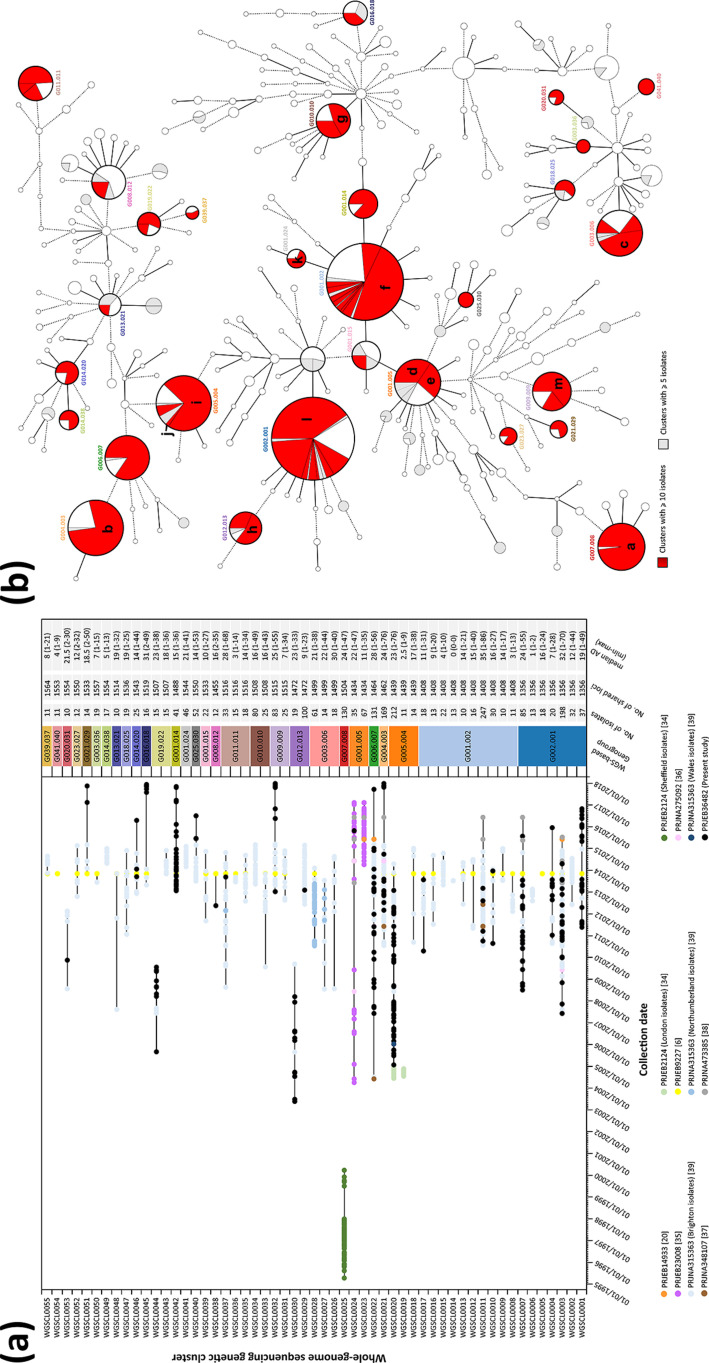
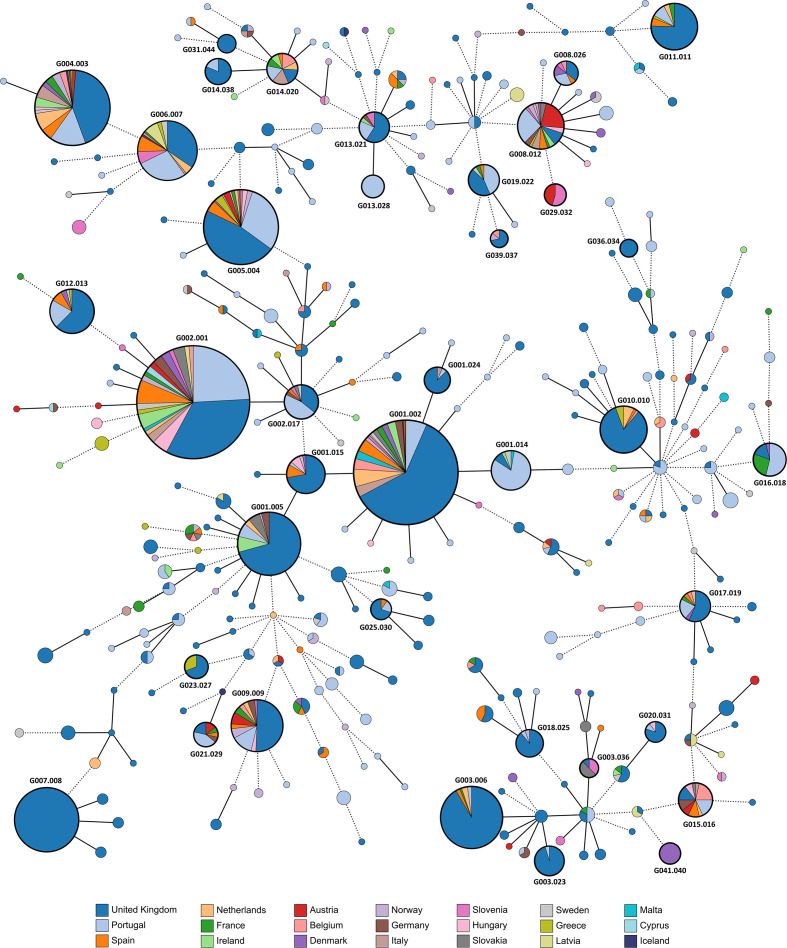
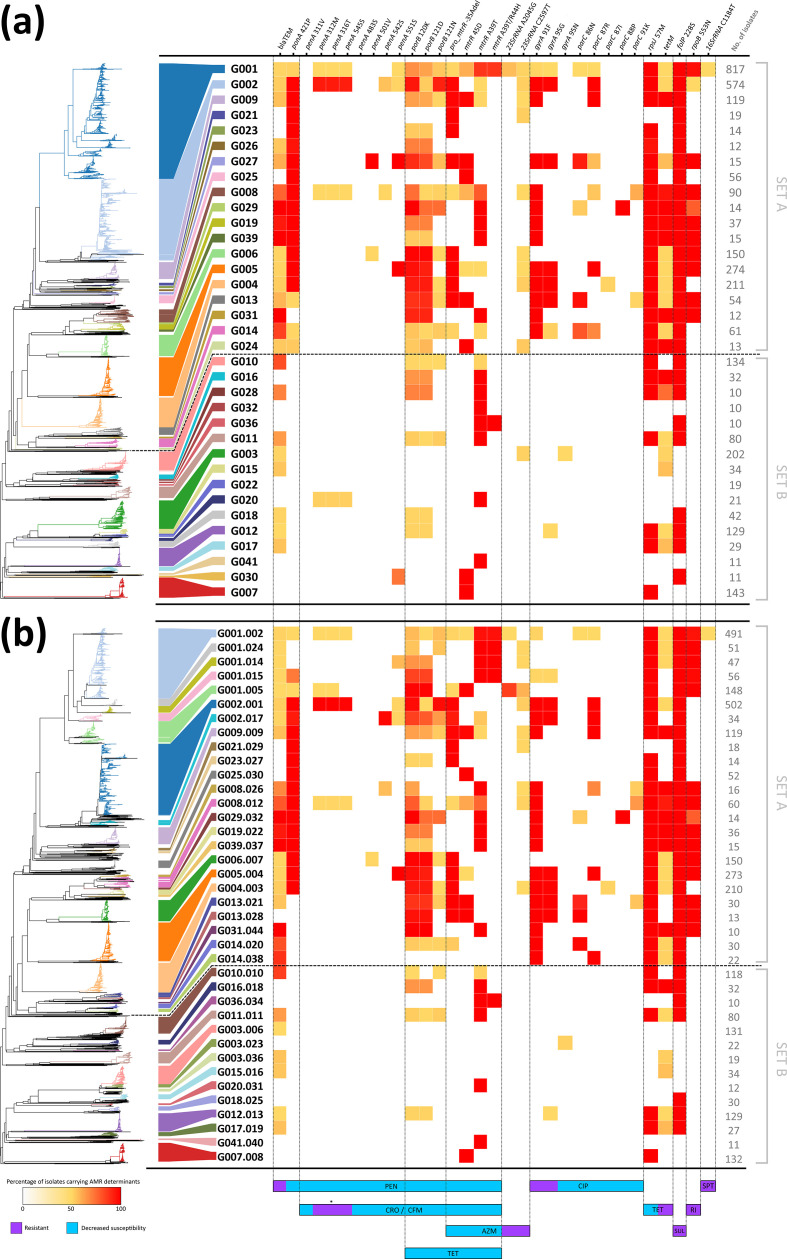
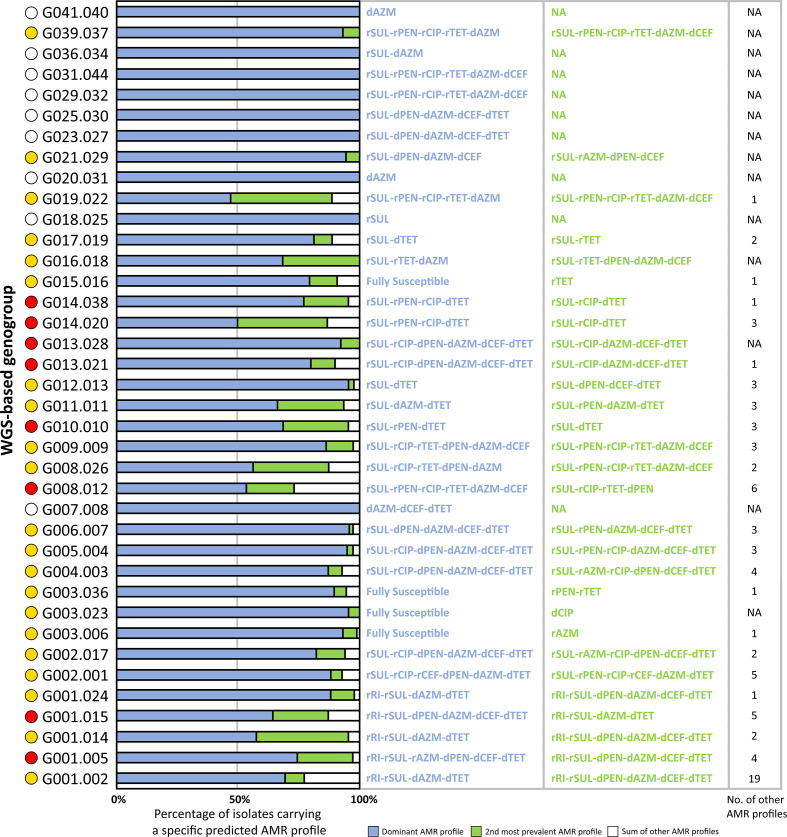
, the bacterium responsible for the sexually transmitted disease gonorrhoea, has shown an extraordinary ability to develop antimicrobial resistance (AMR) to multiple classes of antimicrobials. With no available vaccine, managing infections demands effective preventive measures, antibiotic treatment and epidemiological surveillance. The latter two are progressively being supported by the generation of whole-genome sequencing (WGS) data on behalf of national and international surveillance programmes. In this context, this study aims to perform clustering into genogroups based on WGS data, for enhanced prospective laboratory surveillance. Particularly, it aims to identify the major circulating WGS-genogroups in Europe and to establish a relationship between these and AMR. Ultimately, it enriches public databases by contributing with WGS data from Portuguese isolates spanning 15 years of surveillance. A total of 3791 carefully inspected genomes from isolates collected across Europe were analysed using a gene-by-gene approach (i.e. using cgMLST). Analysis of cluster composition and stability allowed the classification of isolates into a two-step hierarchical genogroup level determined by two allelic distance thresholds revealing cluster stability. Genogroup clustering in general agreed with available typing methods [i.e. MLST (multilocus sequence typing), NG-MAST ( multi-antigen sequence typing) and PubMLST core-genome groups], highlighting the predominant genogroups circulating in Europe, and revealed that the vast majority of the genogroups present a dominant AMR profile. Additionally, a non-static gene-by-gene approach combined with a more discriminatory threshold for potential epidemiological linkage enabled us to match data with previous reports on outbreaks or transmission chains. In conclusion, this genogroup assignment allows a comprehensive analysis of genetic diversity and the identification of the WGS-based genogroups circulating in Europe, while facilitating the assessment (and continuous monitoring) of their frequency, geographical dispersion and potential association with specific AMR signatures. This strategy may benefit public-health actions through the prioritization of genogroups to be controlled, the identification of emerging resistance carriage, and the potential facilitation of data sharing and communication.
淋病奈瑟菌是一种引起性传播疾病的细菌,它对抗微生物药物的多种类别表现出非凡的耐药能力。由于没有可用的疫苗,管理淋病感染需要有效的预防措施、抗生素治疗和流行病学监测。后两者正逐步得到国家和国际监测计划代表的全基因组测序 (WGS) 数据的支持。在这种情况下,本研究旨在根据 WGS 数据进行基因群聚类,以加强前瞻性实验室监测。特别是,它旨在确定欧洲主要的循环 WGS 基因群,并确定这些基因群与抗生素耐药性之间的关系。最终,通过贡献来自葡萄牙 15 年监测的分离株的 WGS 数据,丰富了公共数据库。使用基因对基因方法(即使用 cgMLST)对来自欧洲各地采集的 3791 个精心检查的基因组进行了分析。通过两步聚类分析,根据两个等位基因距离阈值确定了聚类的稳定性,将分离株分为两个层次的基因群。一般来说,基因群聚类与现有的 分型方法(即 MLST(多位点序列分型)、NG-MAST(多抗原序列分型)和 PubMLST 核心基因组群)一致,突出了欧洲循环的主要基因群,并表明目前存在的绝大多数基因群都具有主要的抗生素耐药性特征。此外,非静态基因对基因方法与潜在流行病学关联的更具区分性的阈值相结合,使我们能够将数据与以前关于暴发或传播链的报告相匹配。总之,这种基因群分配允许对遗传多样性进行全面分析,并确定在欧洲循环的基于 WGS 的基因群,同时促进对其频率、地理分布和与特定抗生素耐药性特征的潜在关联的评估(和持续监测)。这种策略可以通过优先控制基因群、识别新兴的耐药性携带以及促进数据共享和交流来使公共卫生行动受益。