Immunology and Oncology, Centro Nacional de Biotecnologia, Madrid, Spain.
Institute of Immunology. Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, Wien, Vienna, Austria.
J Immunother Cancer. 2020 Nov;8(2). doi: 10.1136/jitc-2020-001521.
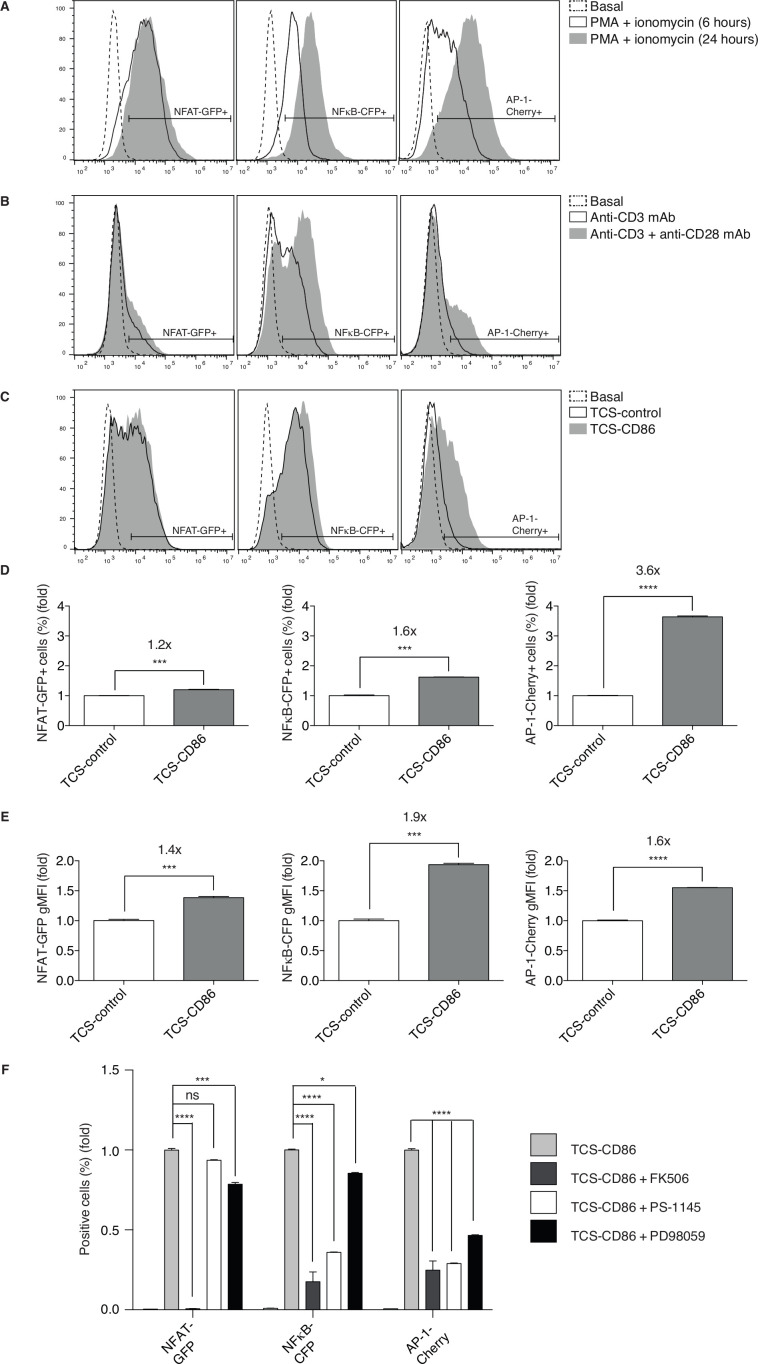
The inhibitory functions triggered by the programmed cell death-1 (PD-1) receptor following binding to its ligand (PD-L1) protect healthy organs from cytotoxic T cells, and neutralize antitumor T cell attack. Antibody-based therapies to block PD-1/PD-L1 interaction have yielded notable results, but most patients eventually develop resistance. This failure is attributed to CD8 T cells achieving hyporesponsive states from which recovery is hardly feasible. Dysfunctional T cell phenotypes are favored by a sustained imbalance in the diacylglycerol (DAG)- and Ca-regulated transcriptional programs. In mice, DAG kinase ζ (DGKζ) facilitates DAG consumption, limiting T cell activation and cytotoxic T cell responses. DGKζ deficiency facilitates tumor rejection in mice without apparent adverse autoimmune effects. Despite its therapeutic potential, little is known about DGKζ function in human T cells, and no known inhibitors target this isoform.
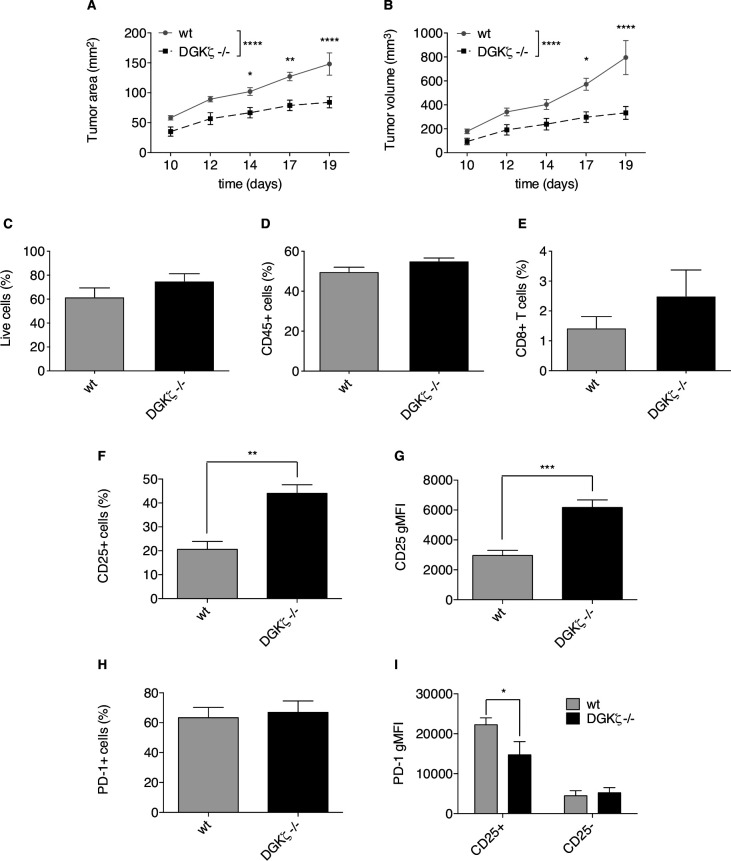
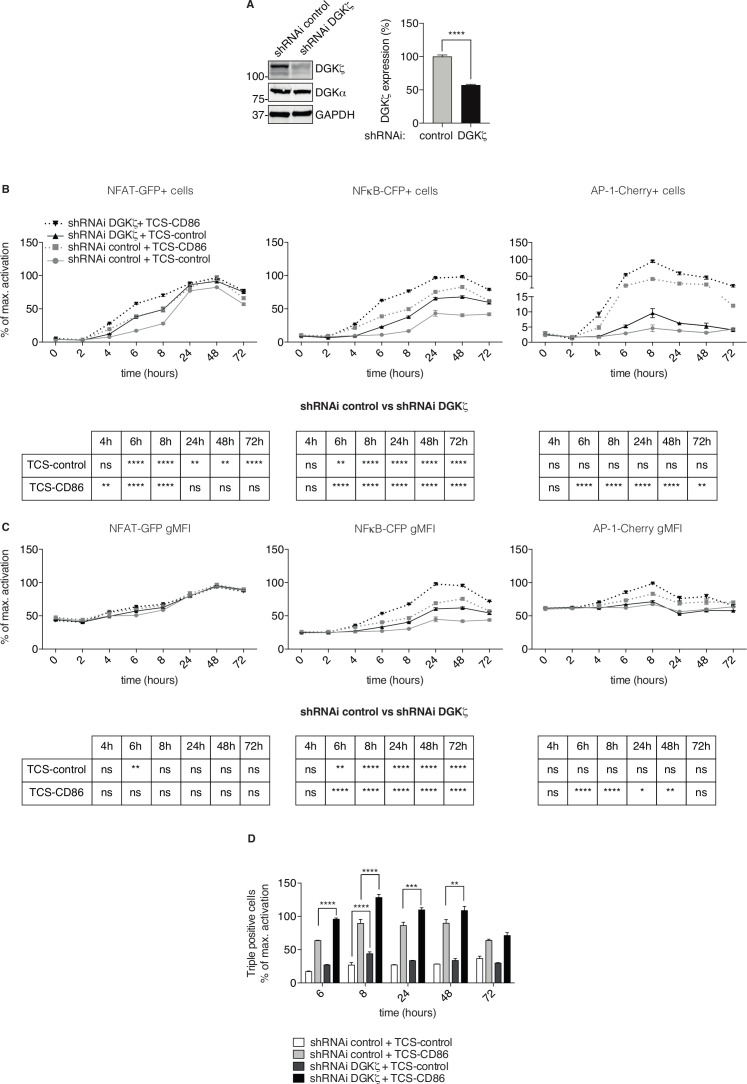
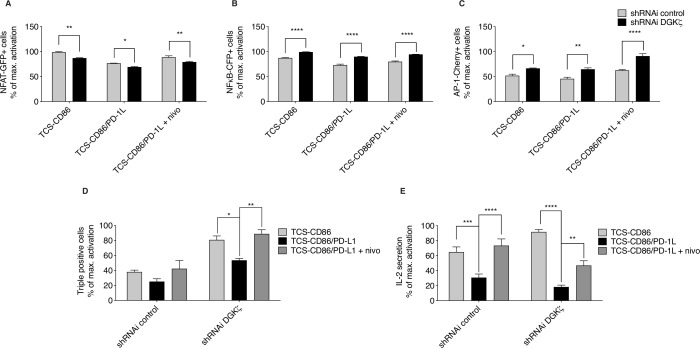
We used a human triple parameter reporter cell line to examine the consequences of DGKζ depletion on the transcriptional restriction imposed by PD-1 ligation. We studied the effect of DGKζ deficiency on PD-1 expression dynamics, as well as the impact of DGKζ absence on the in vivo growth of MC38 adenocarcinoma cells.
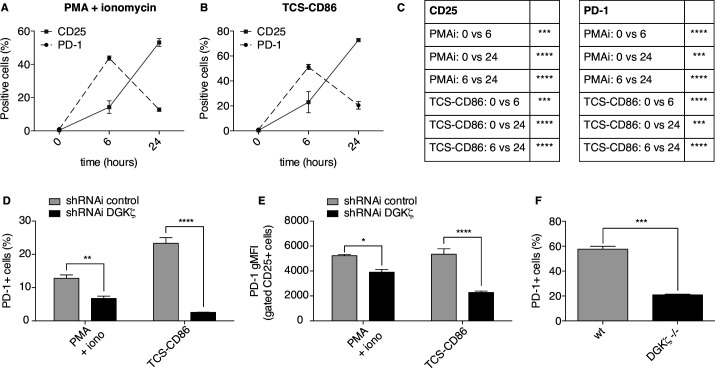
We demonstrate that DGKζ depletion enhances DAG-regulated transcriptional programs, promoting interleukin-2 production and partially counteracting PD-1 inhibitory functions. DGKζ loss results in limited PD-1 expression and enhanced expansion of cytotoxic CD8 T cell populations. This is observed even in immunosuppressive milieus, and correlates with the reduced ability of MC38 adenocarcinoma cells to form tumors in DGKζ-deficient mice.
Our results, which define a role for DGKζ in the control of PD-1 expression, confirm DGKζ potential as a therapeutic target as well as a biomarker of CD8 T cell dysfunctional states.
程序性细胞死亡-1(PD-1)受体与其配体(PD-L1)结合后触发的抑制功能可保护健康器官免受细胞毒性 T 细胞的侵害,并中和抗肿瘤 T 细胞攻击。基于抗体的阻断 PD-1/PD-L1 相互作用的疗法已取得显著效果,但大多数患者最终会产生耐药性。这种失败归因于 CD8 T 细胞处于低反应状态,几乎不可能从中恢复。持续的二酰基甘油(DAG)和 Ca 调节转录程序失衡有利于功能失调的 T 细胞表型。在小鼠中,DAG 激酶 ζ(DGKζ)促进 DAG 消耗,限制 T 细胞激活和细胞毒性 T 细胞反应。DGKζ 缺乏促进小鼠肿瘤排斥,而无明显的自身免疫不良影响。尽管具有治疗潜力,但人们对 DGKζ 在人类 T 细胞中的功能知之甚少,也没有已知的抑制剂针对该同工酶。
我们使用人类三参数报告细胞系来研究 DGKζ 耗竭对 PD-1 连接诱导的转录限制的影响。我们研究了 DGKζ 缺乏对 PD-1 表达动力学的影响,以及 DGKζ 缺失对 MC38 腺癌细胞体内生长的影响。
我们证明 DGKζ 耗竭增强了 DAG 调节的转录程序,促进了白细胞介素-2 的产生,并部分抵消了 PD-1 的抑制功能。DGKζ 缺失导致 PD-1 表达受限,细胞毒性 CD8 T 细胞群体扩增受限。即使在免疫抑制环境中也观察到这种情况,并且与 MC38 腺癌细胞在 DGKζ 缺陷小鼠中形成肿瘤的能力降低相关。
我们的研究结果定义了 DGKζ 在 PD-1 表达控制中的作用,证实了 DGKζ 作为治疗靶点以及 CD8 T 细胞功能失调状态的生物标志物的潜力。