Department of Pharmacology and Toxicology, Faculty of Medicine, American University of Beirut, Riad El-Solh 1107 2020, Beirut 11-0236, Lebanon.
Rafic Hariri School of Nursing, American University of Beirut, Riad El-Solh 1107 2020, Beirut 11-0236, Lebanon.
Int J Mol Sci. 2020 Nov 27;21(23):9005. doi: 10.3390/ijms21239005.
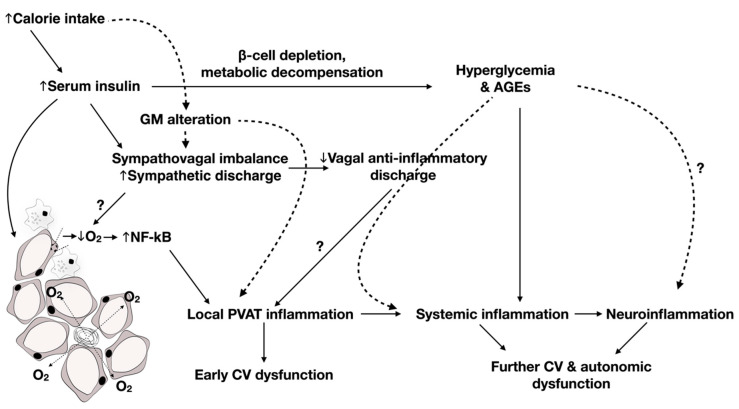
Cardiac autonomic neuropathy (CAN) is one of the earliest complications of type 2 diabetes (T2D), presenting a silent cause of cardiovascular morbidity and mortality. Recent research relates the pathogenesis of cardiovascular disease in T2D to an ensuing chronic, low-grade proinflammatory and pro-oxidative environment, being the hallmark of the metabolic syndrome. Metabolic inflammation emerges as adipose tissue inflammatory changes extending systemically, on the advent of hyperglycemia, to reach central regions of the brain. In light of changes in glucose and insulin homeostasis, dysbiosis or alteration of the gut microbiome (GM) emerges, further contributing to inflammatory processes through increased gut and blood-brain barrier permeability. Interestingly, studies reveal that the determinants of oxidative stress and inflammation progression exist at the crossroad of CAN manifestations, dictating their evolution along the natural course of T2D development. Indeed, sympathetic and parasympathetic deterioration was shown to correlate with markers of adipose, vascular, and systemic inflammation. Additionally, evidence points out that dysbiosis could promote a sympatho-excitatory state through differentially affecting the secretion of hormones and neuromodulators, such as norepinephrine, serotonin, and γ-aminobutyric acid, and acting along the renin-angiotensin-aldosterone axis. Emerging neuronal inflammation and concomitant autophagic defects in brainstem nuclei were described as possible underlying mechanisms of CAN in experimental models of metabolic syndrome and T2D. Drugs with anti-inflammatory characteristics provide potential avenues for targeting pathways involved in CAN initiation and progression. The aim of this review is to delineate the etiology of CAN in the context of a metabolic disorder characterized by elevated oxidative and inflammatory load.
心脏自主神经病变(CAN)是 2 型糖尿病(T2D)最早的并发症之一,是心血管发病率和死亡率的一个无声原因。最近的研究将 T2D 中心血管疾病的发病机制与随之而来的慢性、低度炎症和氧化应激环境联系起来,这是代谢综合征的标志。代谢炎症是由于高血糖导致脂肪组织炎症变化向全身扩展,并延伸到大脑中枢区域。鉴于葡萄糖和胰岛素稳态的变化,肠道微生物组(GM)的失调或改变出现,通过增加肠道和血脑屏障通透性进一步促进炎症过程。有趣的是,研究表明,氧化应激和炎症进展的决定因素存在于 CAN 表现的交叉路口,决定了它们在 T2D 发展的自然过程中的演变。事实上,交感神经和副交感神经的恶化与脂肪、血管和系统炎症的标志物相关。此外,有证据表明,通过不同地影响激素和神经调节剂(如去甲肾上腺素、血清素和γ-氨基丁酸)的分泌,肠道菌群失调可能会通过促进交感兴奋状态来促进炎症,并且沿着肾素-血管紧张素-醛固酮轴发挥作用。在代谢综合征和 T2D 的实验模型中,描述了脑干核中神经元炎症和同时发生的自噬缺陷作为 CAN 的可能潜在机制。具有抗炎特性的药物为针对 CAN 起始和进展所涉及的途径提供了潜在途径。本综述的目的是在以氧化应激和炎症负荷升高为特征的代谢紊乱背景下阐述 CAN 的病因。