Institute for Cellular and Molecular Biology, Department of Molecular Biosciences, University of Texas, Austin, Texas, USA.
Institute for Cellular and Molecular Biology, Department of Molecular Biosciences, University of Texas, Austin, Texas, USA.
J Biol Chem. 2021 Jan-Jun;296:100143. doi: 10.1074/jbc.RA120.016617. Epub 2020 Dec 10.
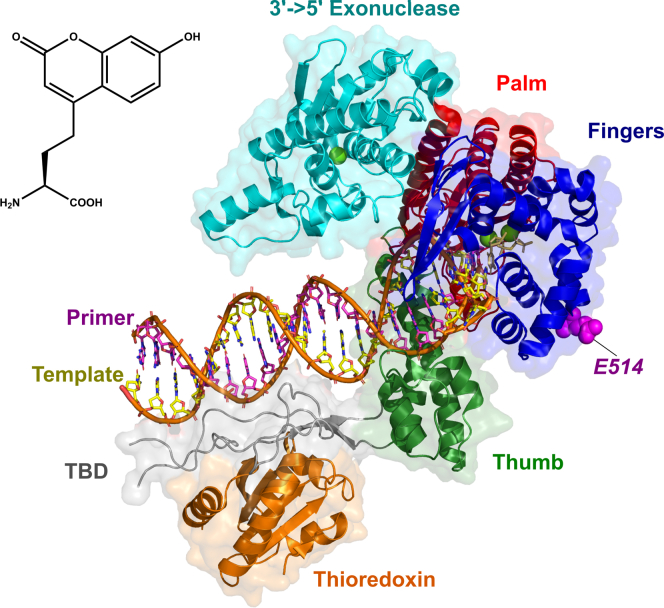
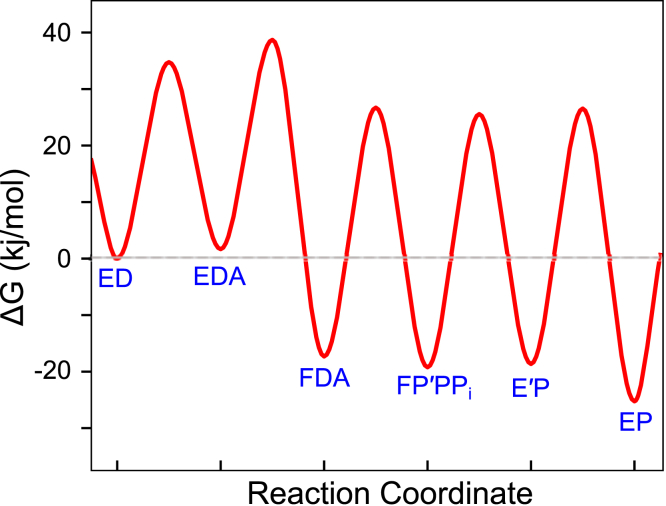
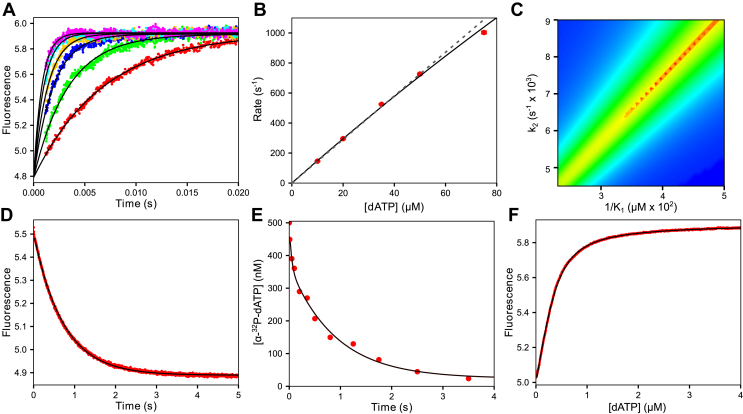
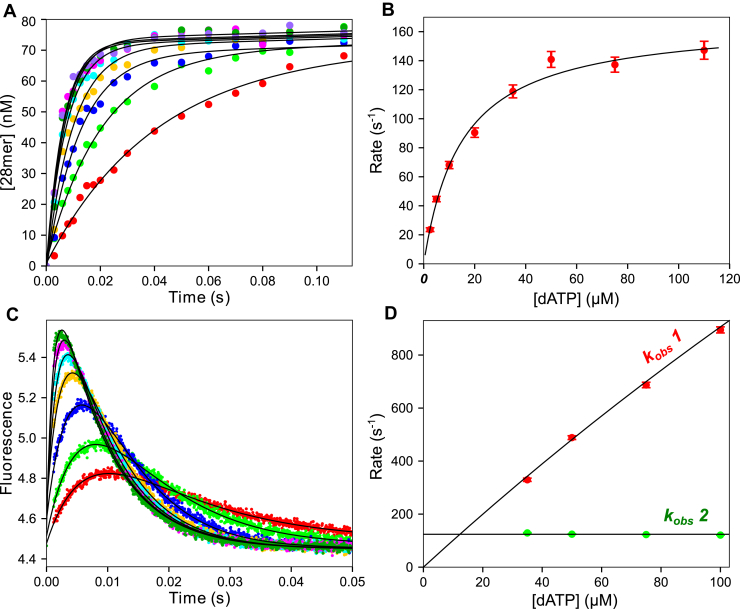
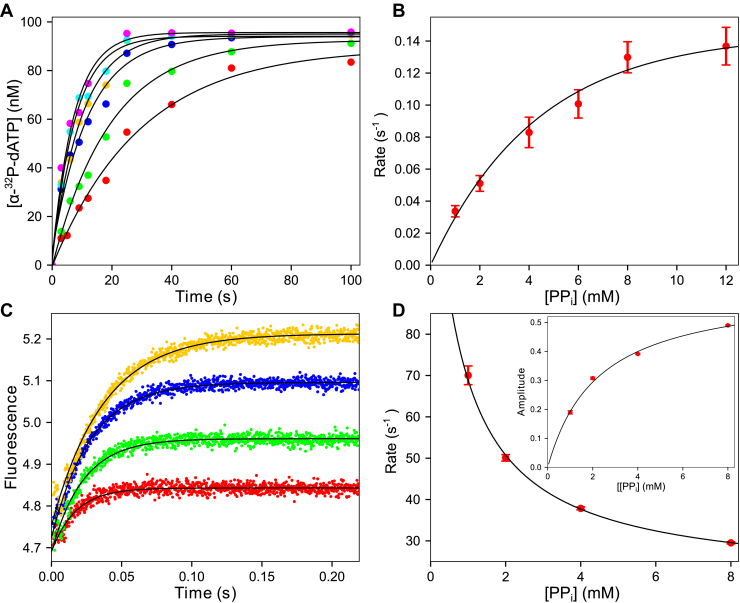
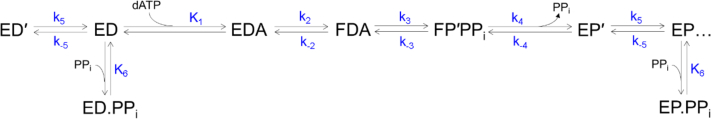
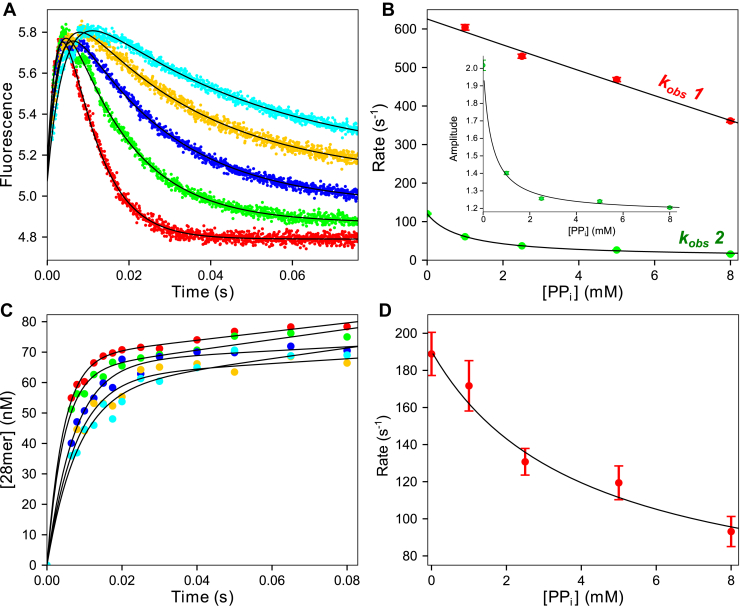
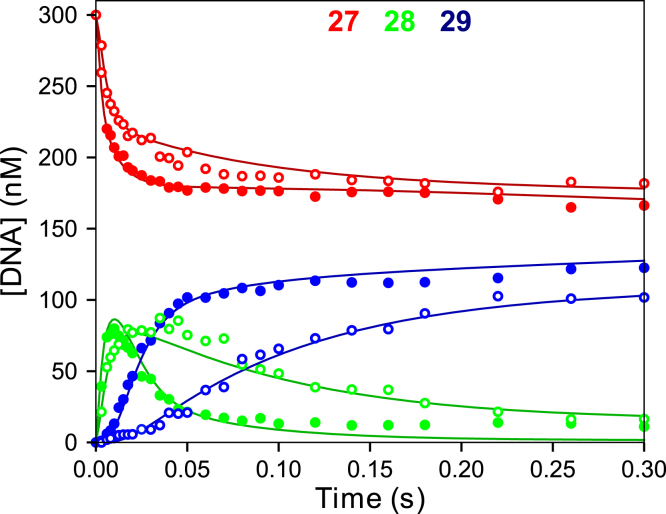
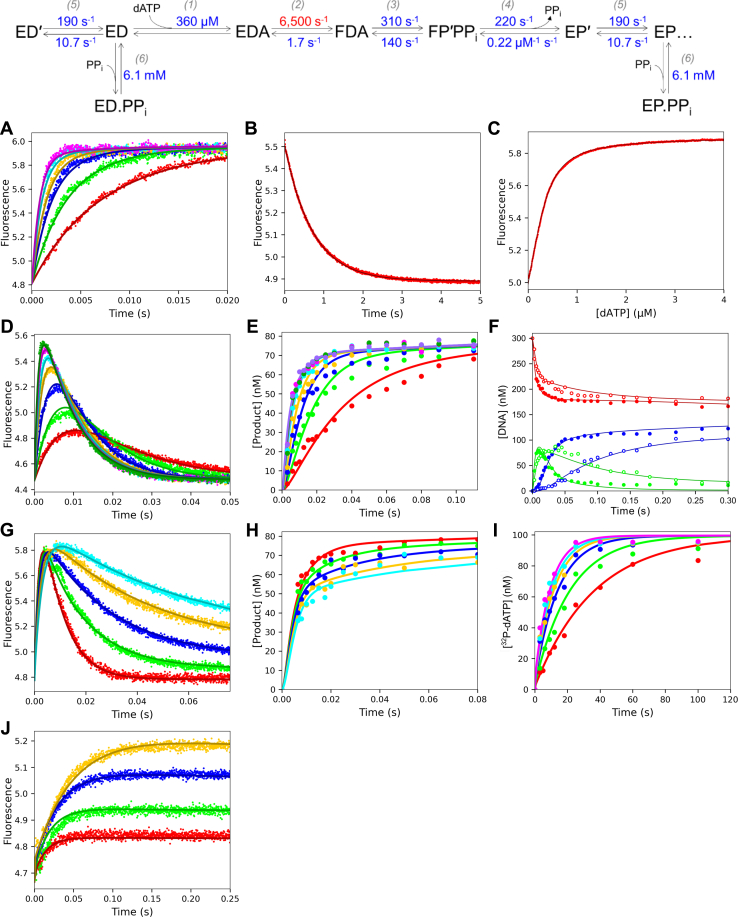
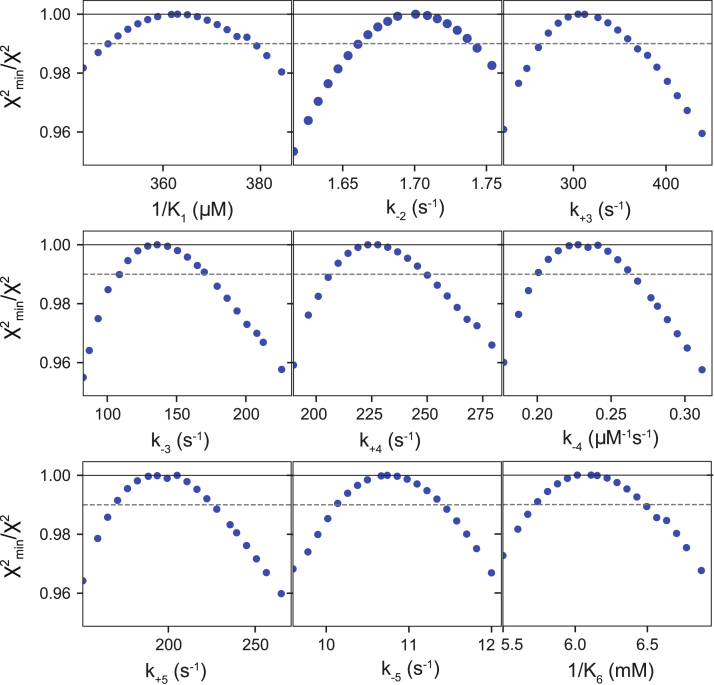
We address the role of enzyme conformational dynamics in specificity for a high-fidelity DNA polymerase responsible for genome replication. We present the complete characterization of the conformational dynamics during the correct nucleotide incorporation forward and reverse reactions using stopped-flow and rapid-quench methods with a T7 DNA polymerase variant containing a fluorescent unnatural amino acid, (7-hydroxy-4-coumarin-yl) ethylglycine, which provides a signal for enzyme conformational changes. We show that the forward conformational change (>6000 s) is much faster than chemistry (300 s) while the enzyme opening to allow release of bound nucleotide (1.7 s) is much slower than chemistry. These parameters show that the conformational change selects a correct nucleotide for incorporation through an induced-fit mechanism. We also measured conformational changes occurring after chemistry and during pyrophosphorolysis, providing new insights into processive polymerization. Pyrophosphorolysis occurs via a conformational selection mechanism as the pyrophosphate binds to a rare pretranslocation state of the enzyme-DNA complex. Global data fitting was achieved by including experiments in the forward and reverse directions to correlate conformational changes with chemical reaction steps. This analysis provided well-constrained values for nine rate constants to establish a complete free-energy profile including the rates of DNA translocation during processive synthesis. Translocation does not follow Brownian ratchet or power stroke models invoking nucleotide binding as the driving force. Rather, translocation is rapid and thermodynamically favorable after enzyme opening and pyrophosphate release, and it appears to limit the rate of processive synthesis at 4 °C.
我们研究了负责基因组复制的高保真 DNA 聚合酶的酶构象动力学在特异性中的作用。我们使用含有荧光非天然氨基酸(7-羟基-4-香豆素基)乙基甘氨酸的 T7 DNA 聚合酶变体,通过停流和快速淬火方法,全面描述了正确核苷酸掺入正向和反向反应过程中的构象动力学。我们表明,正向构象变化(>6000 s)比化学反应(300 s)快得多,而酶打开以允许结合核苷酸释放(1.7 s)比化学反应慢得多。这些参数表明,构象变化通过诱导契合机制选择正确的核苷酸进行掺入。我们还测量了化学变化后和焦磷酸解过程中发生的构象变化,为连续聚合提供了新的见解。焦磷酸解通过构象选择机制发生,因为焦磷酸根结合到酶-DNA 复合物的罕见预迁移状态。通过包括正向和反向实验来关联构象变化与化学反应步骤,实现了全局数据拟合。这一分析为建立包括连续合成过程中 DNA 易位速率在内的完整自由能谱提供了 9 个速率常数的良好约束值。易位不遵循布朗棘轮或功率冲程模型,将核苷酸结合作为驱动力。相反,酶打开和焦磷酸根释放后易位迅速且热力学有利,并且似乎在 4°C 时限制了连续合成的速率。