Department of Applied Mathematics, Western University, London, Ontario, Canada.
Department of Ecology and Evolution, Yale University, New Haven, Connecticut, USA.
PLoS Comput Biol. 2020 Dec 4;16(12):e1008482. doi: 10.1371/journal.pcbi.1008482. eCollection 2020 Dec.
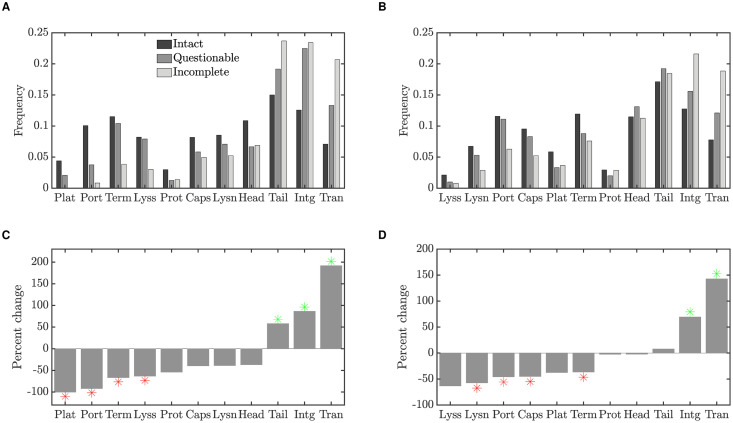
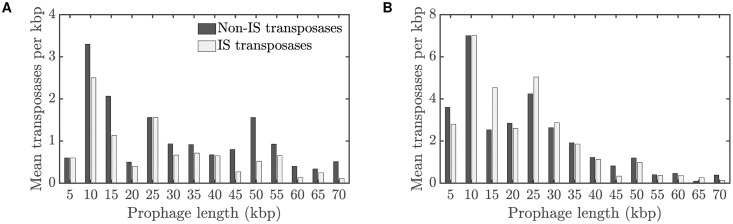
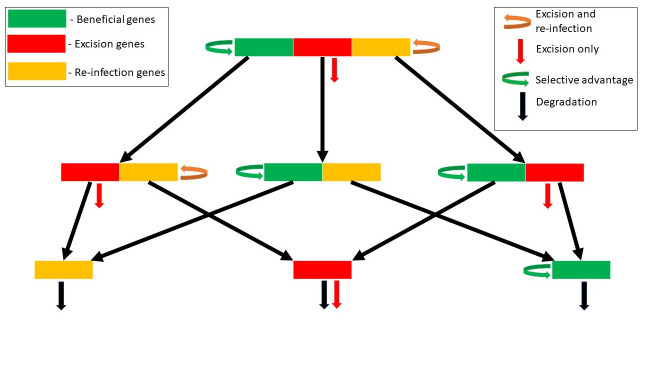
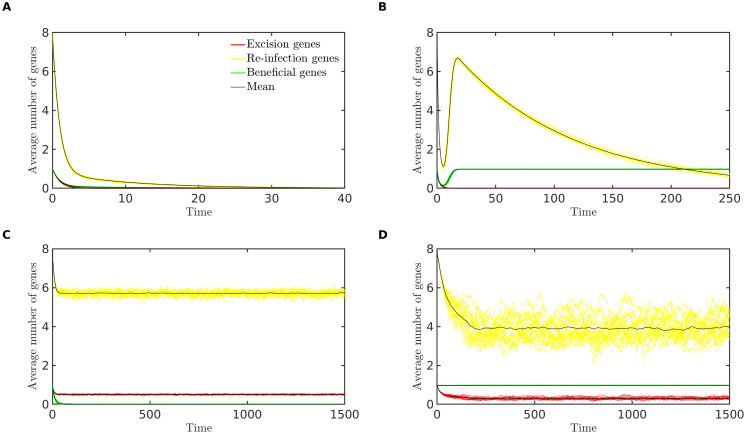
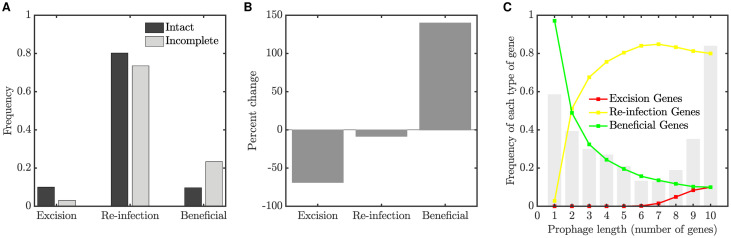
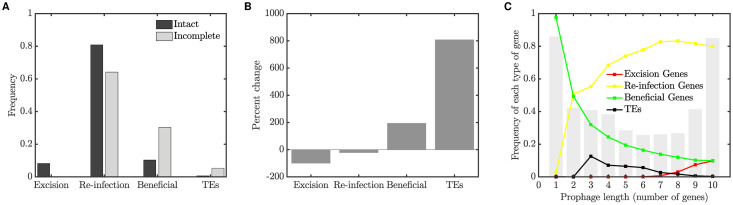
Integrated into their bacterial hosts' genomes, prophage sequences exhibit a wide diversity of length and gene content, from highly degraded cryptic sequences to intact, functional prophages that retain a full complement of lytic-function genes. We apply three approaches-bioinformatics, analytical modelling and computational simulation-to understand the diverse gene content of prophages. In the bioinformatics work, we examine the distributions of over 50,000 annotated prophage genes identified in 1384 prophage sequences, comparing the gene repertoires of intact and incomplete prophages. These data indicate that genes involved in the replication, packaging, and release of phage particles have been preferentially lost in incomplete prophages, while tail fiber, transposase and integrase genes are significantly enriched. Consistent with these results, our mathematical and computational approaches predict that genes involved in phage lytic function are preferentially lost, resulting in shorter prophages that often retain genes that benefit the host. Informed by these models, we offer novel hypotheses for the enrichment of integrase and transposase genes in cryptic prophages. Overall, we demonstrate that functional and cryptic prophages represent a diversity of genetic sequences that evolve along a parasitism-mutualism continuum.
整合到它们的细菌宿主基因组中,噬菌体序列表现出广泛的长度和基因组成多样性,从高度退化的隐秘序列到完整的、保留全套裂解功能基因的功能性噬菌体。我们应用三种方法——生物信息学、分析建模和计算模拟——来理解噬菌体的多样基因组成。在生物信息学工作中,我们研究了在 1384 个噬菌体序列中鉴定出的超过 50000 个注释噬菌体基因的分布,比较了完整和不完整噬菌体的基因库。这些数据表明,参与噬菌体粒子复制、包装和释放的基因在不完整噬菌体中已被优先丢失,而尾部纤维、转座酶和整合酶基因则显著富集。与这些结果一致,我们的数学和计算方法预测,与噬菌体裂解功能相关的基因更易丢失,导致噬菌体变短,而通常保留对宿主有益的基因。受这些模型的启发,我们为隐秘噬菌体中整合酶和转座酶基因的富集提出了新的假说。总的来说,我们证明了功能性和隐秘噬菌体代表了沿着寄生-共生连续体进化的多样化遗传序列。