Ingine Inc. Cleveland Ohio USA and the Dirac Foundation, Oxfordshire, UK.
Comput Biol Med. 2021 Jan;128:104124. doi: 10.1016/j.compbiomed.2020.104124. Epub 2020 Nov 21.
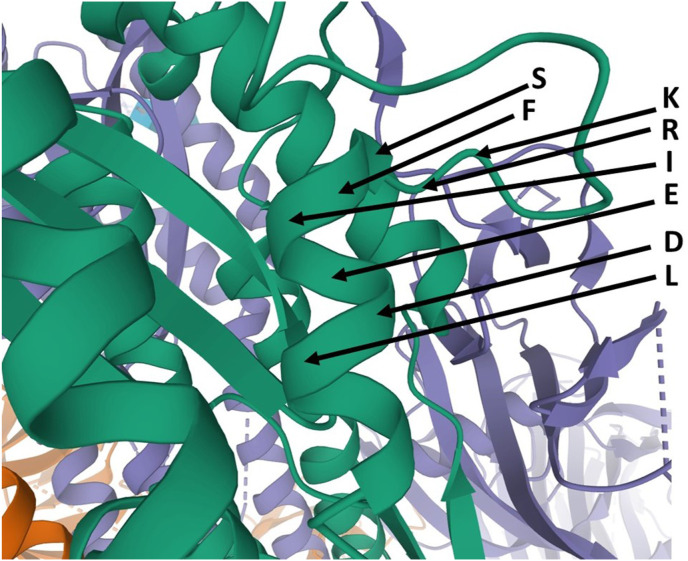
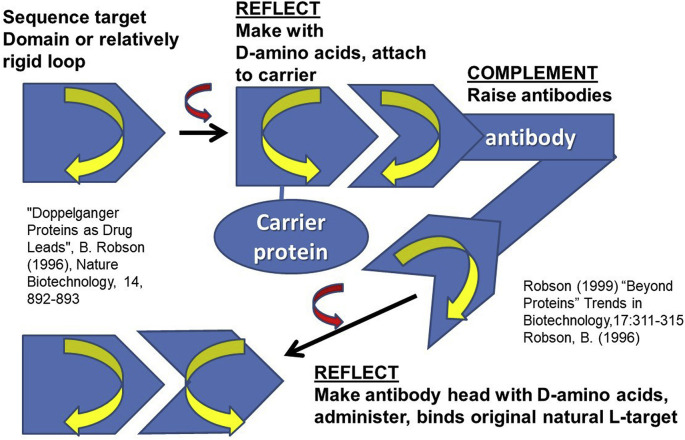
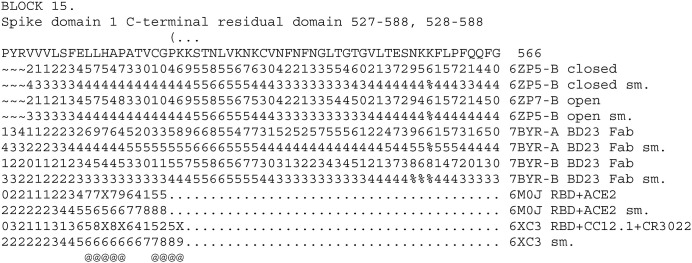
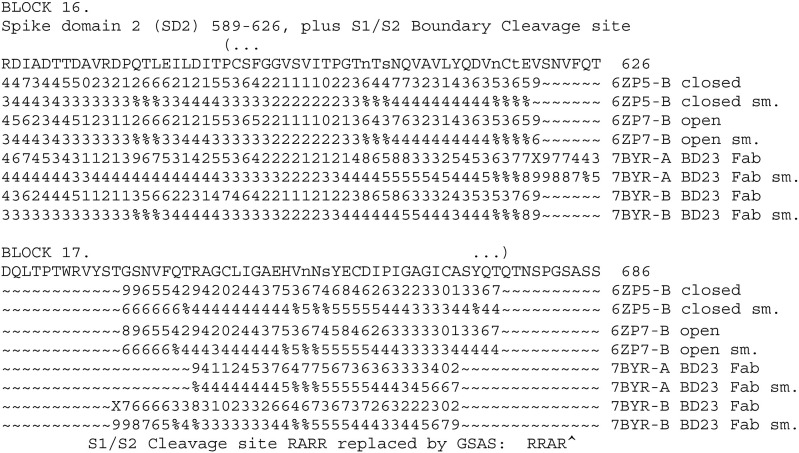
The aim of the present study is to discuss the design of peptide vaccines and peptidomimetics against SARS-COV-2, to develop and apply a method of protein structure analysis that is particularly appropriate to applying and discussing such design, and also to use that method to summarize some important features of the SARS-COV-2 spike protein sequence. A tool for assessing sidechain exposure in the SARS-CoV-2 spike glycoprotein is described. It extends to assessing accessibility of sidechains by considering several different three-dimensional structure determinations of SARS-CoV-2 and SARS-CoV-1 spike protein. The method is designed to be insensitive to a distance limit for counting neighboring atoms and the results are in good agreement with the physical chemical properties and exposure trends of the 20 naturally occurring sidechains. The spike protein sequence is analyzed with comment regarding exposable character. It includes studies of complexes with antibody elements and ACE2. These indicate changes in exposure at sites remote to those at which the antibody binds. They are of interest concerning design of synthetic peptide vaccines, and for peptidomimetics as a basis of drug discovery. The method was also developed in order to provide linear (one-dimensional) information that can be used along with other bioinformatics data of this kind in data mining and machine learning, potentially as genomic data regarding protein polymorphisms to be combined with more traditional clinical data.
本研究旨在讨论针对 SARS-CoV-2 的肽疫苗和类肽设计,开发并应用一种特别适用于应用和讨论此类设计的蛋白质结构分析方法,并且还使用该方法总结 SARS-CoV-2 刺突蛋白序列的一些重要特征。描述了一种评估 SARS-CoV-2 刺突糖蛋白侧链暴露的工具。它通过考虑 SARS-CoV-2 和 SARS-CoV-1 刺突蛋白的几种不同三维结构测定来扩展到评估侧链的可及性。该方法旨在对计数相邻原子的距离限制不敏感,并且结果与 20 种天然存在的侧链的物理化学性质和暴露趋势非常吻合。对刺突蛋白序列进行了分析,并对可暴露特征进行了评论。它包括对与抗体元素和 ACE2 结合的复合物的研究。这些研究表明,在与抗体结合的部位之外的部位的暴露发生了变化。它们在设计合成肽疫苗和作为药物发现的肽模拟物方面具有重要意义。该方法还被开发出来,以便提供线性(一维)信息,可以与这种其他类型的生物信息学数据一起用于数据挖掘和机器学习,可能作为与更传统的临床数据相结合的关于蛋白质多态性的基因组数据。