SAMRC/NHLS/UCT Molecular Mycobacteriology Research Unit & DST/NRF Centre of Excellence for Biomedical TB Research, Institute of Infectious Disease and Molecular Medicine & Department of Pathology, University of Cape Town, Cape Town, South Africa.
H3D Drug Discovery and Development Centre, Department of Chemistry, University of Cape Town, Cape Town, South Africa.
Front Cell Infect Microbiol. 2020 Nov 13;10:582416. doi: 10.3389/fcimb.2020.582416. eCollection 2020.
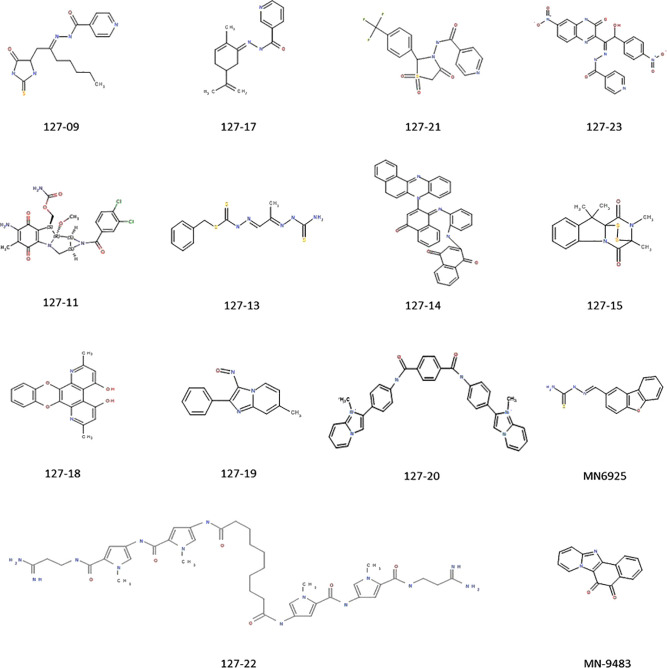
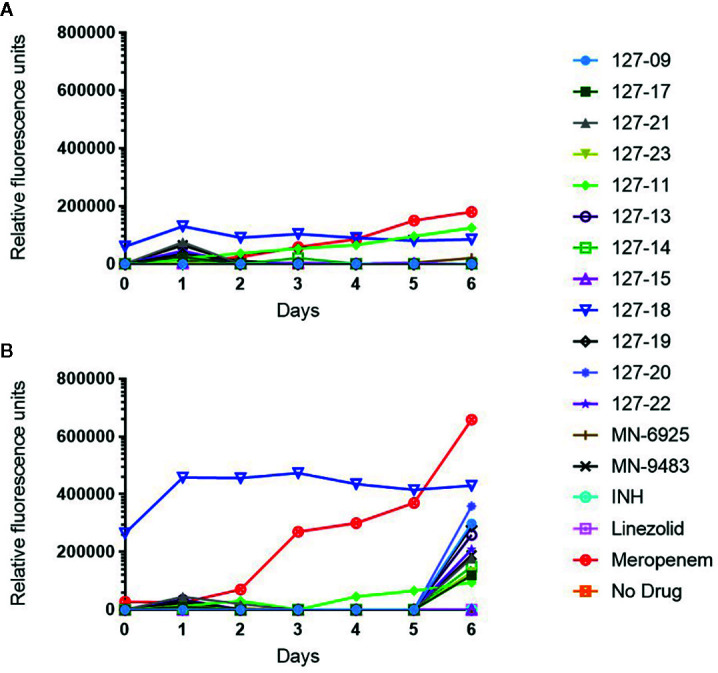
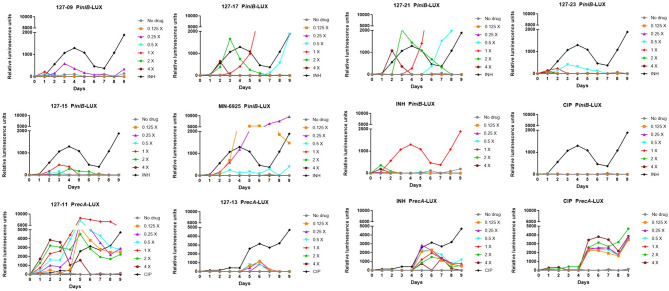
Compounds with novel modes of action are urgently needed to develop effective combination therapies for the treatment of tuberculosis. In this study, a series of compounds was evaluated for activity against replicating and Vero cell line toxicity. Fourteen of the compounds with activities in the low micrometer range and a favorable selectivity index were classified using reporter strains of which showed that six interfered with cell wall metabolism and one disrupted DNA metabolism. Counter-screening against strains carrying mutations in promiscuous drug targets argued against DprE1 and MmpL3 as hits of any of the cell wall actives and eliminated the cytochrome complex as a target of any of the compounds. Instead, whole-genome sequencing of spontaneous resistant mutants and/or counter-screening against common isoniazid-resistant mutants of revealed that four of the six cell wall-active compounds, all pyridine carboxamide analogues, were metabolized by KatG to form InhA inhibitors. Resistance to two of these compounds was associated with mutations in that did not confer cross-resistance to isoniazid. Of the remaining seven compounds, low-level resistance to one was associated with an inactivating mutation in Rv0678, the regulator of the MmpS5-MmpL5 system, which has been implicated in non-specific efflux of multiple chemotypes. Another mapped to the mycothiol-dependent reductase, Rv2466c, suggesting a prodrug mechanism of action in that case. The inability to isolate spontaneous resistant mutants to the seven remaining compounds suggests that they act mechanisms which have yet to be elucidated.
需要具有新型作用模式的化合物来开发有效的结核病联合疗法。在这项研究中,评估了一系列化合物对复制期结核分枝杆菌和 Vero 细胞系毒性的活性。14 种具有低微摩尔范围活性和良好选择性指数的化合物被分类为报告菌株,结果表明其中 6 种化合物干扰细胞壁代谢,1 种化合物破坏 DNA 代谢。针对携带多种药物靶点突变株的反向筛选表明,DprE1 和 MmpL3 不是任何细胞壁活性化合物的靶点,细胞色素 复合物也不是任何化合物的靶点。相反,自发耐药突变体的全基因组测序和/或针对结核分枝杆菌常见异烟肼耐药突变体的反向筛选表明,6 种细胞壁活性化合物中的 4 种,均为吡啶甲酰胺类似物,被 KatG 代谢形成 InhA 抑制剂。其中两种化合物的耐药性与不引起异烟肼交叉耐药的 突变有关。在其余的 7 种化合物中,一种化合物的低水平耐药性与 MmpS5-MmpL5 系统的调节剂 Rv0678 的失活突变有关,该突变已被牵连到多种化学型的非特异性外排。另一种化合物与依赖于分枝菌硫醇的还原酶 Rv2466c 有关,表明在这种情况下存在前药作用机制。无法分离到对其余 7 种化合物的自发耐药突变体,表明它们作用于尚未阐明的作用机制。