Xu Jingya, Fu Yuhua, Hu Yan, Yin Lilin, Tang Zhenshuang, Yin Dong, Zhu Mengjin, Yu Mei, Li Xinyun, Zhou Yang, Zhao Shuhong, Liu Xiaolei
Key Laboratory of Agricultural Animal Genetics, Breeding and Reproduction, Ministry of Education & Key Laboratory of Swine Genetics and Breeding, Ministry of Agriculture & College of Animal Science and Technology, Huazhong Agricultural University, Wuhan, 430070, Hubei, PR China.
J Anim Sci Biotechnol. 2020 Dec 3;11(1):115. doi: 10.1186/s40104-020-00520-8.
A large number of pig breeds are distributed around the world, their features and characteristics vary among breeds, and they are valuable resources. Understanding the underlying genetic mechanisms that explain across-breed variation can help breeders develop improved pig breeds.
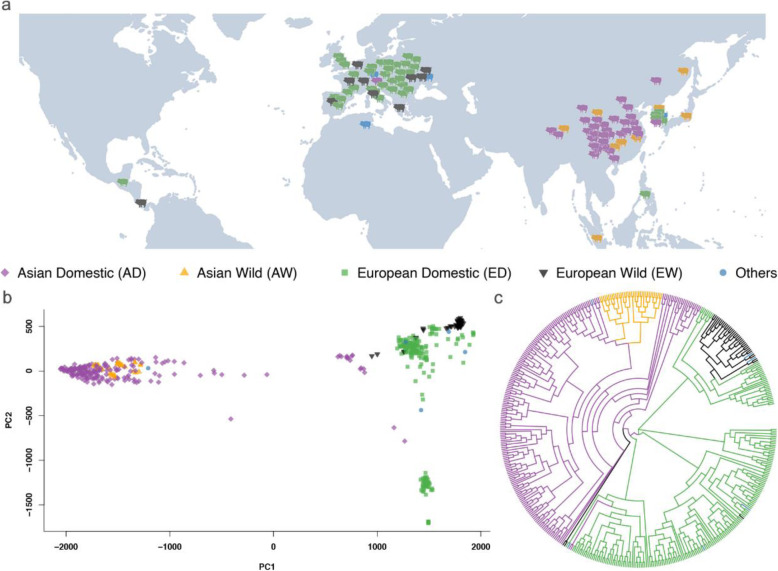
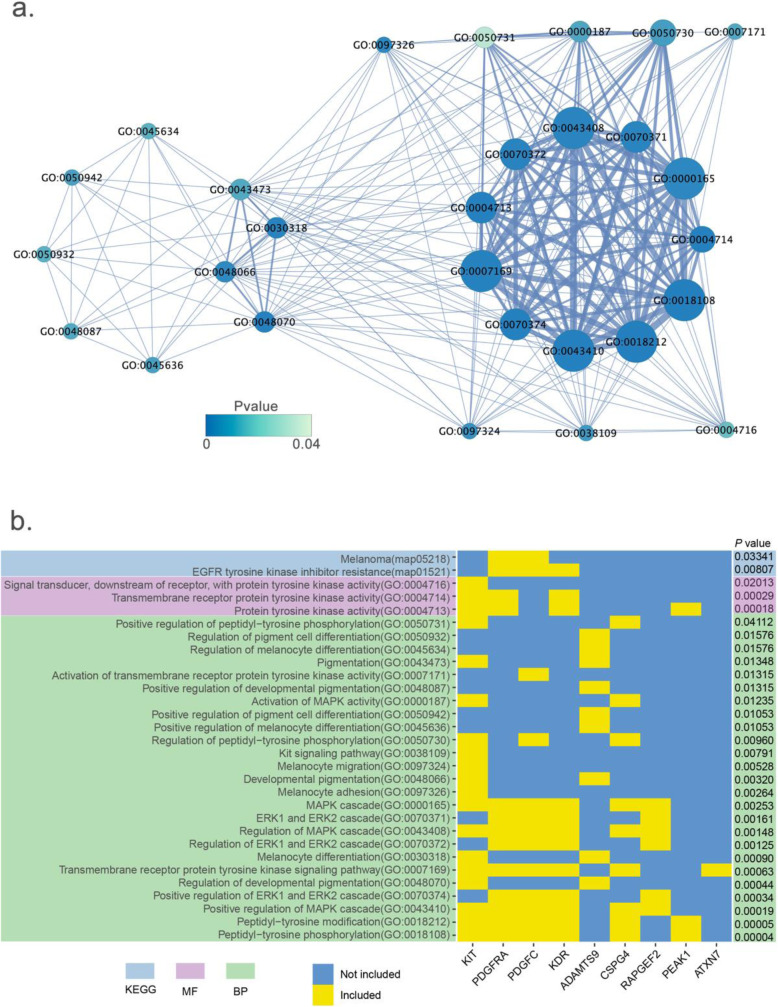
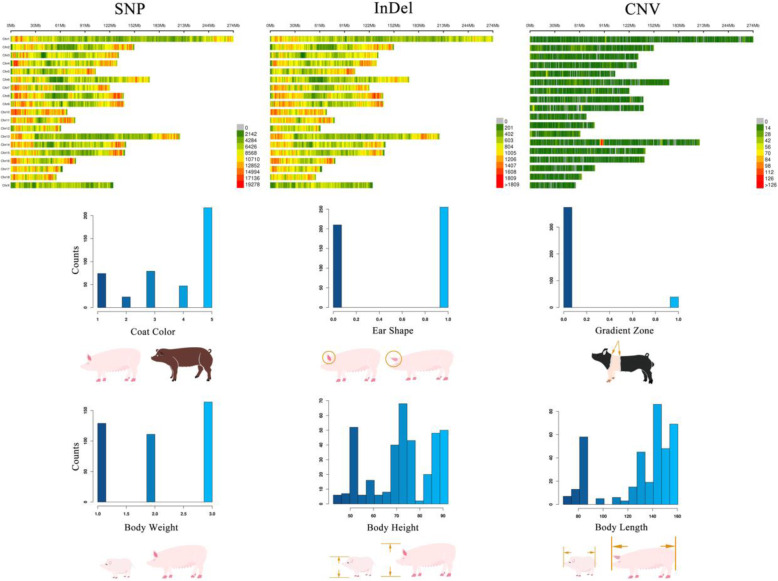
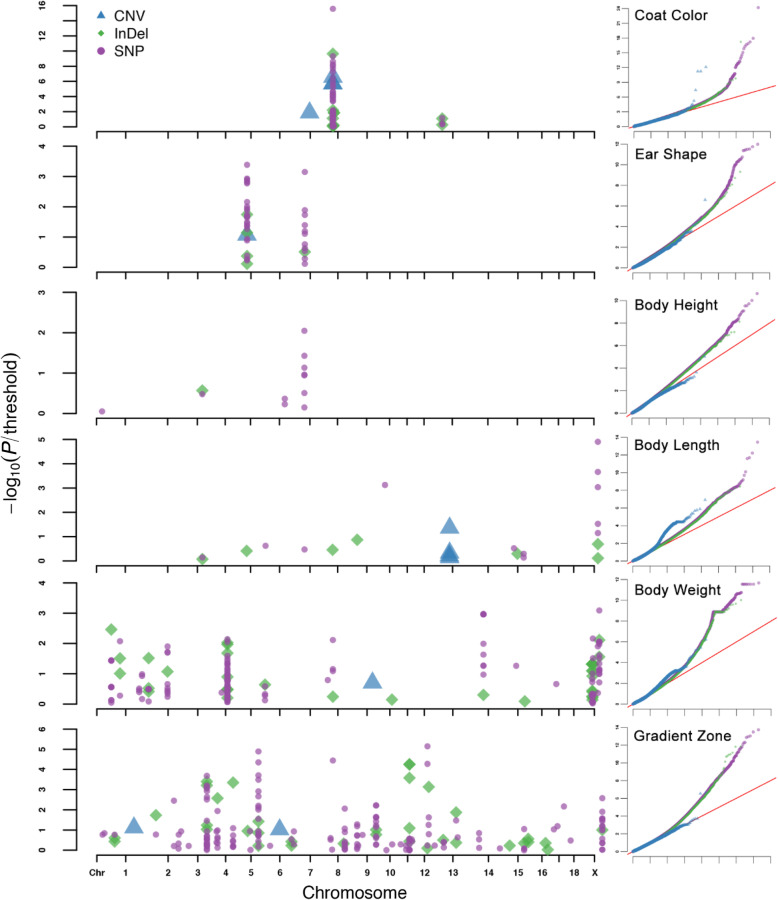
In this study, we performed GWAS using a standard mixed linear model with three types of genome variants (SNP, InDel, and CNV) that were identified from public, whole-genome, sequencing data sets. We used 469 pigs of 57 breeds, and we identified and analyzed approximately 19 million SNPs, 1.8 million InDels, and 18,016 CNVs. We defined six biological phenotypes by the characteristics of breed features to identify the associated genome variants and candidate genes, which included coat color, ear shape, gradient zone, body weight, body length, and body height. A total of 37 candidate genes was identified, which included 27 that were reported previously (e.g., PLAG1 for body weight), but the other 10 were newly detected candidate genes (e.g., ADAMTS9 for coat color).
Our study indicated that using GWAS across a modest number of breeds with high density genome variants provided efficient mapping of complex traits.
世界范围内分布着大量猪品种,各品种的特征和特性存在差异,它们都是宝贵的资源。了解解释品种间差异的潜在遗传机制有助于育种者培育改良猪品种。
在本研究中,我们使用标准混合线性模型对从公开的全基因组测序数据集中鉴定出的三种类型的基因组变异(单核苷酸多态性、插入缺失和拷贝数变异)进行全基因组关联研究。我们使用了57个品种的469头猪,鉴定并分析了约1900万个单核苷酸多态性、180万个插入缺失和18016个拷贝数变异。我们根据品种特征定义了六种生物学表型,以鉴定相关的基因组变异和候选基因,包括毛色、耳形、渐变区、体重、体长和体高。共鉴定出37个候选基因,其中27个是先前报道过的(如与体重相关的PLAG1),但另外10个是新检测到的候选基因(如与毛色相关的ADAMTS9)。
我们的研究表明,使用高密度基因组变异对数量适中的品种进行全基因组关联研究能够有效地定位复杂性状。