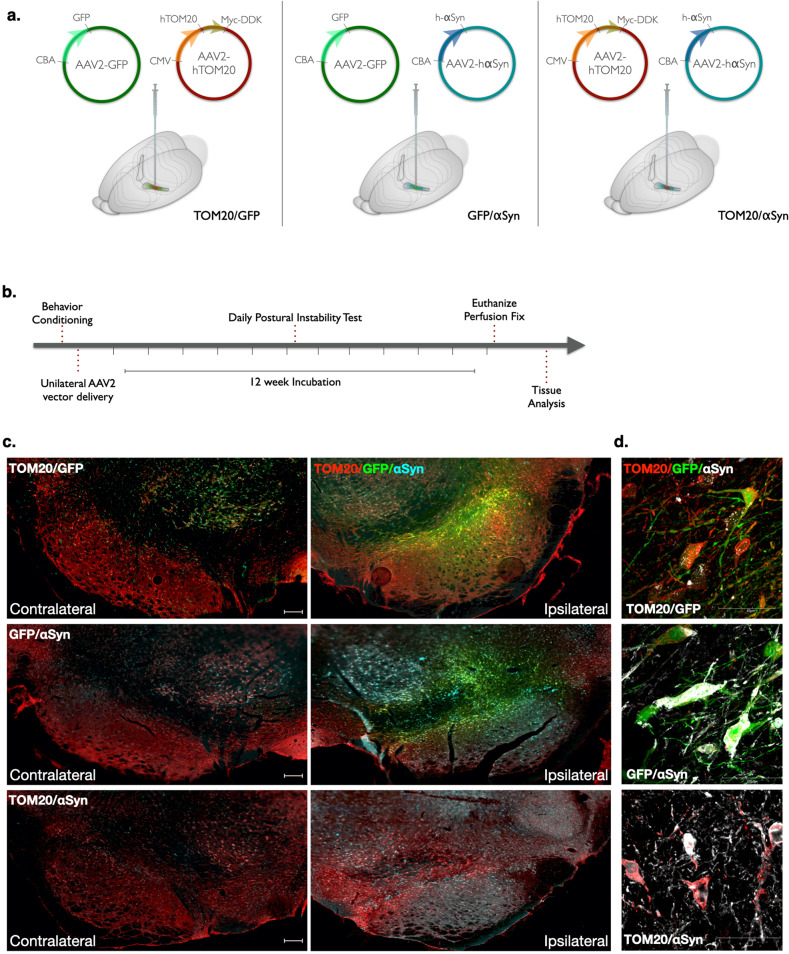
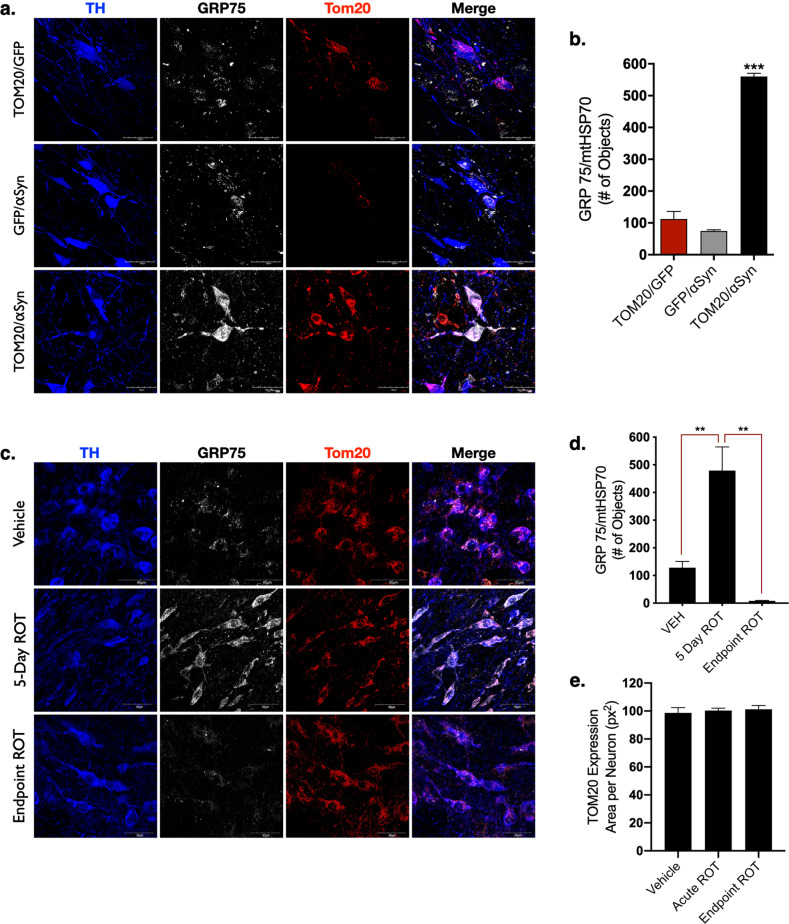
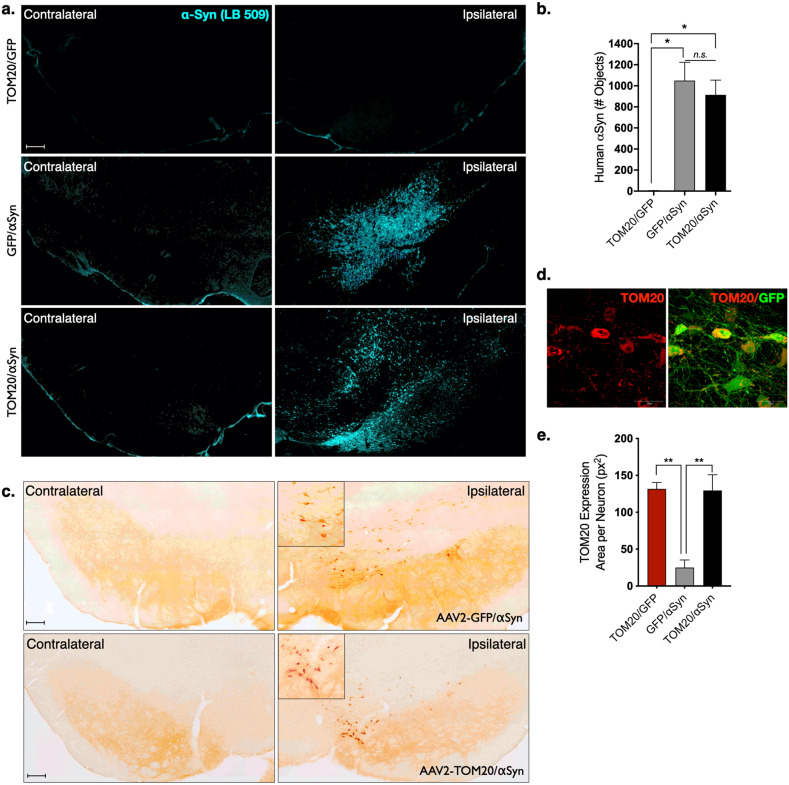
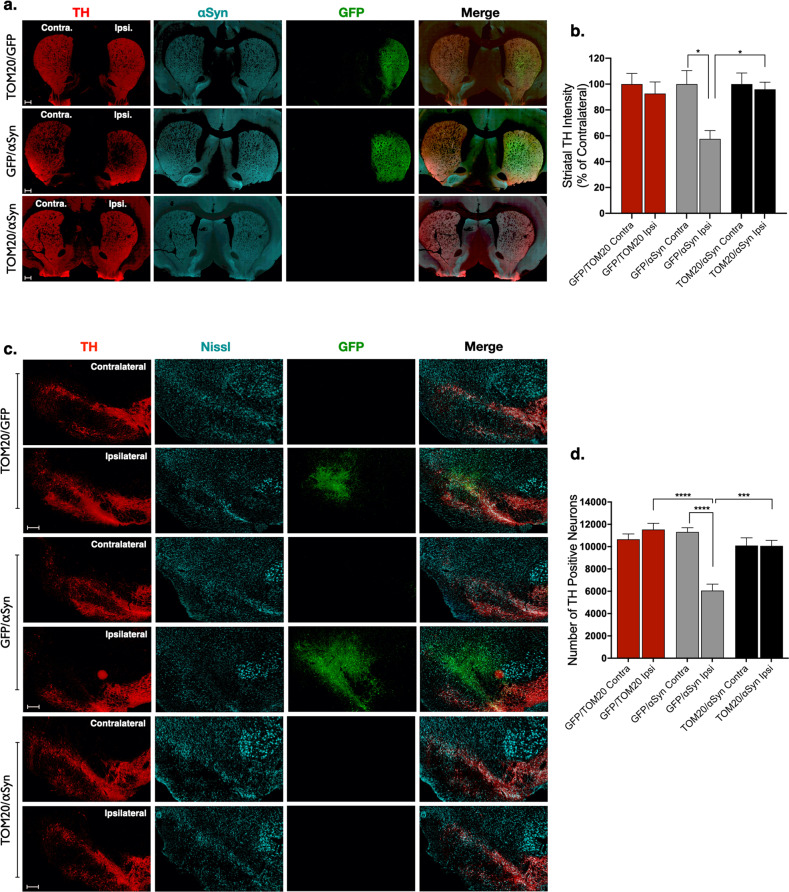
De Miranda Briana R, Rocha Emily M, Castro Sandra L, Greenamyre J Timothy
Pittsburgh Institute for Neurodegenerative Diseases and Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA.
NPJ Parkinsons Dis. 2020 Dec 8;6(1):38. doi: 10.1038/s41531-020-00139-6.
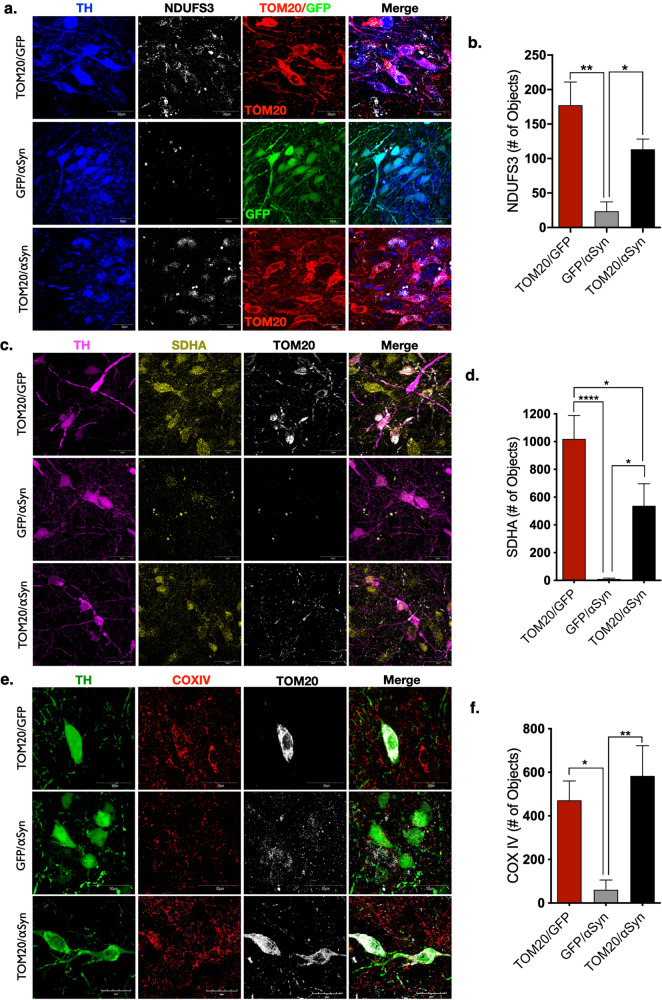
Dopaminergic neurons of the substantia nigra are selectively vulnerable to mitochondrial dysfunction, which is hypothesized to be an early and fundamental pathogenic mechanism in Parkinson's disease (PD). Mitochondrial function depends on the successful import of nuclear-encoded proteins, many of which are transported through the TOM20-TOM22 outer mitochondrial membrane import receptor machinery. Recent data suggests that post-translational modifications of α-synuclein promote its interaction with TOM20 at the outer mitochondrial membrane and thereby inhibit normal protein import, leading to dysfunction, and death of dopaminergic neurons. As such, preservation of mitochondrial import in the face of α-synuclein accumulation might be a strategy to prevent dopaminergic neurodegeneration, however, this is difficult to assess using current in vivo models of PD. To this end, we established an exogenous co-expression system, utilizing AAV2 vectors to overexpress human α-synuclein and TOM20, individually or together, in the adult Lewis rat substantia nigra to assess whether TOM20 overexpression attenuates α-synuclein-induced dopaminergic neurodegeneration. Twelve weeks after viral injection, we observed that AAV2-TOM20 expression was sufficient to prevent loss of nigral dopaminergic neurons caused by AAV2-αSyn overexpression. The observed TOM20-mediated dopaminergic neuron preservation appeared to be due, in part, to the rescued expression (and presumed import) of nuclear-encoded mitochondrial electron transport chain proteins that were inhibited by α-synuclein overexpression. In addition, TOM20 overexpression rescued the expression of the chaperone protein GRP75/mtHSP70/mortalin, a stress-response protein involved in α-synuclein-induced injury. Collectively, these data indicate that TOM20 expression prevents α-synuclein-induced mitochondrial dysfunction, which is sufficient to rescue dopaminergic neurons in the adult rat brain.
黑质中的多巴胺能神经元对线粒体功能障碍具有选择性易损性,据推测这是帕金森病(PD)早期的一个基本致病机制。线粒体功能取决于核编码蛋白的成功导入,其中许多蛋白通过TOM20-TOM22线粒体外膜导入受体机制进行转运。最近的数据表明,α-突触核蛋白的翻译后修饰促进其在线粒体外膜与TOM20相互作用,从而抑制正常的蛋白导入,导致多巴胺能神经元功能障碍和死亡。因此,在α-突触核蛋白积累的情况下维持线粒体导入可能是预防多巴胺能神经退行性变的一种策略,然而,使用当前的PD体内模型很难对此进行评估。为此,我们建立了一个外源性共表达系统,利用腺相关病毒2型(AAV2)载体在成年Lewis大鼠黑质中单独或共同过表达人α-突触核蛋白和TOM20,以评估TOM20过表达是否能减轻α-突触核蛋白诱导的多巴胺能神经退行性变。病毒注射12周后,我们观察到AAV2-TOM20的表达足以预防由AAV2-αSyn过表达引起的黑质多巴胺能神经元丢失。观察到的TOM20介导的多巴胺能神经元保护作用似乎部分归因于被α-突触核蛋白过表达抑制的核编码线粒体电子传递链蛋白的表达恢复(以及推测的导入)。此外,TOM20过表达恢复了伴侣蛋白GRP75/mtHSP70/ mortalin的表达,GRP75/mtHSP70/ mortalin是一种参与α-突触核蛋白诱导损伤的应激反应蛋白。总体而言,这些数据表明TOM20的表达可预防α-突触核蛋白诱导的线粒体功能障碍,这足以挽救成年大鼠脑中的多巴胺能神经元。