Puusepp Sanna, Reinson Karit, Pajusalu Sander, van Kuilenburg André B P, Dobritzsch Doreen, Roelofsen Jeroen, Stenzel Werner, Õunap Katrin
Department of Clinical Genetics, United Laboratories, Tartu University Hospital, Tartu, Estonia.
Department of Clinical Genetics, Institute of Clinical Medicine, Faculty of Medicine, University of Tartu, Tartu, Estonia.
Mol Genet Metab Rep. 2020 Nov 18;25:100677. doi: 10.1016/j.ymgmr.2020.100677. eCollection 2020 Dec.
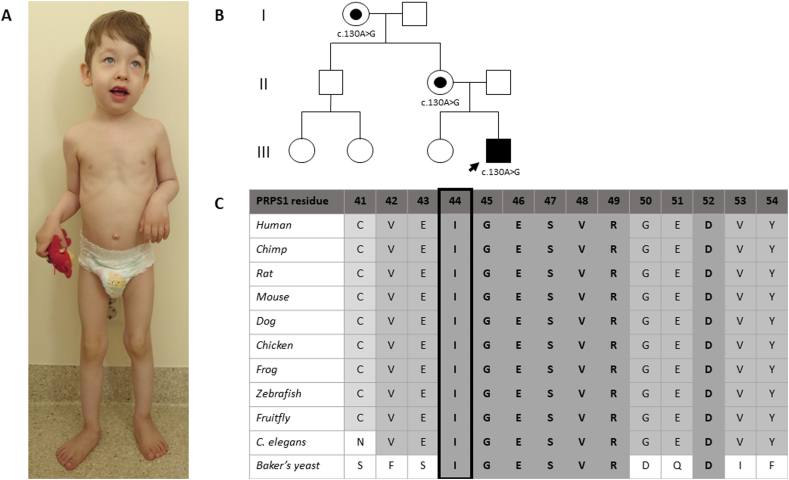
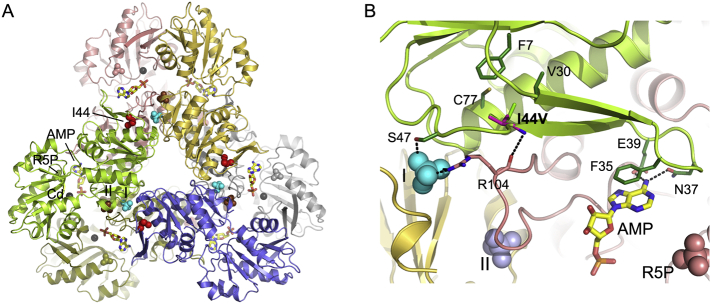
The gene, located on Xq22.3, encodes phosphoribosyl-pyrophosphate synthetase (PRPS), a key enzyme in de novo purine synthesis. Three clinical phenotypes are associated with loss-of-function variants and decreased PRPS activity: Arts syndrome (OMIM: 301835), Charcot-Marie-Tooth disease type 5 (CMTX5, OMIM: 311070), and nonsyndromic X-linked deafness (DFN2, OMIM: 304500). Hearing loss is present in all cases. CMTX5 patients also show peripheral neuropathy and optic atrophy. Arts syndrome includes developmental delay, intellectual disability, ataxia, and susceptibility to infections, in addition to the above three features. Gain-of-function variants result in PRPS superactivity (OMIM: 300661) with hyperuricemia and gout. We report a 6-year-old boy who presented with marked generalized muscular hypotonia, global developmental delay, lack of speech, trunk instability, exercise intolerance, hypomimic face with open mouth, oropharyngeal dysphagia, dysarthria, and frequent upper respiratory tract infections. However, his nerve conduction velocity, audiologic, and funduscopic investigations were normal. A novel hemizygous variant, c.130A > G p.(Ile44Val), was found in the gene by panel sequencing. PRPS activity in erythrocytes was markedly reduced, confirming the pathogenicity of the variant. Serum uric acid and urinary purine and pyrimidine metabolite levels were normal. In conclusion, we present a novel loss-of-function variant in a patient with some clinical features of Arts syndrome, but lacking a major attribute, hearing loss, which is congenital/early-onset in all other reported Arts syndrome patients. In addition, it is important to acknowledge that normal levels of serum and urinary purine and pyrimidine metabolites do not exclude -related disorders.
该基因位于Xq22.3,编码磷酸核糖焦磷酸合成酶(PRPS),这是嘌呤从头合成中的关键酶。功能丧失变异和PRPS活性降低与三种临床表型相关:阿茨综合征(OMIM:301835)、5型夏科-马里-图斯病(CMTX5,OMIM:311070)和非综合征性X连锁耳聋(DFN2,OMIM:304500)。所有病例均有听力损失。CMTX5患者还表现出周围神经病变和视神经萎缩。除上述三个特征外,阿茨综合征还包括发育迟缓、智力残疾、共济失调和易感染。功能获得变异导致PRPS活性过强(OMIM:300661),伴有高尿酸血症和痛风。我们报告一名6岁男孩,表现为明显的全身肌肉张力减退、全面发育迟缓、无语言能力、躯干不稳、运动不耐受、面无表情且张口、口咽吞咽困难、构音障碍和频繁的上呼吸道感染。然而,他的神经传导速度、听力和眼底检查均正常。通过基因panel测序在该基因中发现了一个新的半合子变异,c.130A>G p.(Ile44Val)。红细胞中的PRPS活性明显降低,证实了该变异的致病性。血清尿酸以及尿嘌呤和嘧啶代谢物水平正常。总之,我们在一名具有阿茨综合征某些临床特征但缺乏一个主要特征即听力损失(在所有其他报道的阿茨综合征患者中为先天性/早发性)的患者中发现了一种新的功能丧失变异。此外,必须认识到血清和尿嘌呤及嘧啶代谢物水平正常并不能排除相关疾病。