Amrute-Nayak Mamta, Pegoli Gloria, Holler Tim, Lopez-Davila Alfredo Jesus, Lanzuolo Chiara, Nayak Arnab
Institute of Molecular and Cell Physiology, Hannover Medical School, Hannover, Germany.
Institute of Biomedical Technologies, National Research Council, Milan, Italy.
J Cachexia Sarcopenia Muscle. 2021 Feb;12(1):159-176. doi: 10.1002/jcsm.12645. Epub 2020 Dec 10.
Chemotherapy is the first line of treatment for cancer patients. However, the side effects cause severe muscle atrophy or chemotherapy-induced cachexia. Previously, the NF-κB/MuRF1-dependent pathway was shown to induce chemotherapy-induced cachexia. We hypothesized that acute collateral toxic effects of chemotherapy on muscles might involve other unknown pathways promoting chemotherapy-induced muscle atrophy. In this study, we investigated differential effects of chemotherapeutic drugs and probed whether alternative molecular mechanisms lead to cachexia.
We employed mouse satellite stem cell-derived primary muscle cells and mouse C2C12 progenitor cell-derived differentiated myotubes as model systems to test the effect of drugs. The widely used chemotherapeutic drugs, such as daunorubicin (Daun), etoposide (Etop), and cytarabine (Ara-C), were tested. Molecular mechanisms by which drug affects the muscle cell organization at epigenetic, transcriptional, and protein levels were measured by employing chromatin immunoprecipitations, endogenous gene expression profiling, co-immunoprecipitation, complementation assays, and confocal microscopy. Myotube function was examined using the electrical stimulation of myotubes to monitor contractile ability (excitation-contraction coupling) post drug treatment.
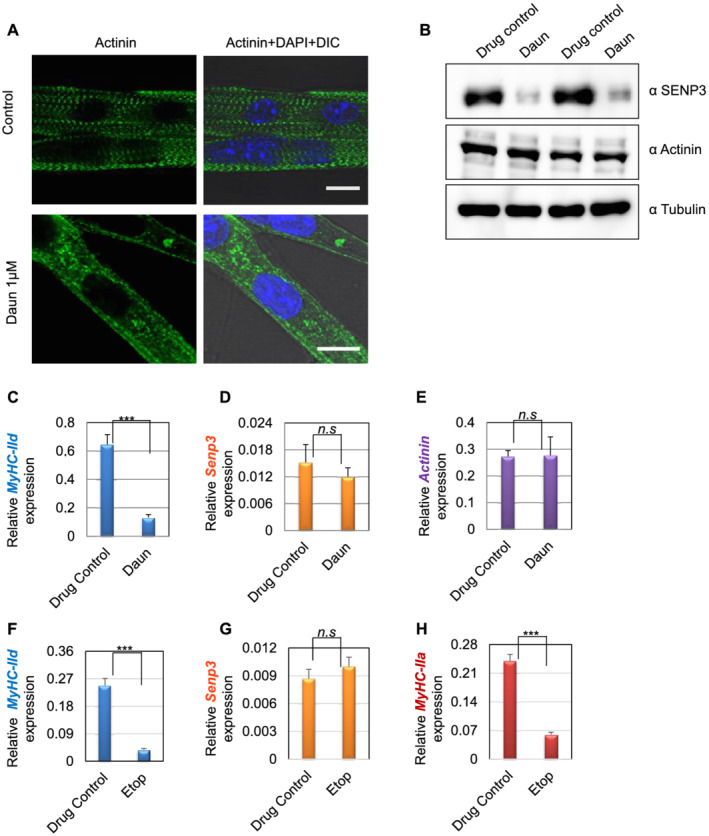
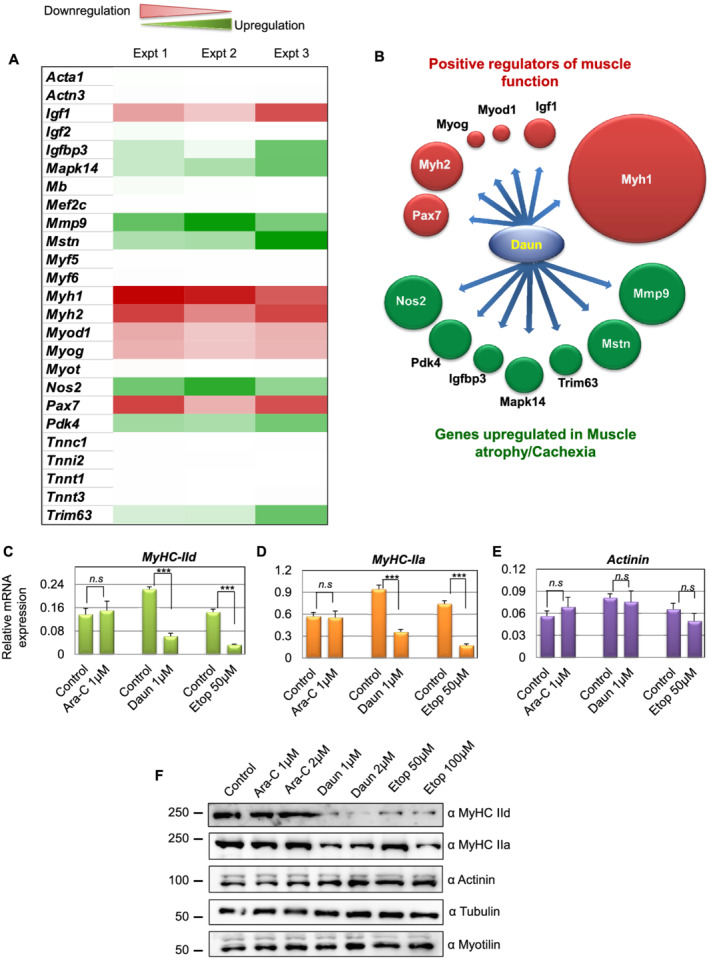
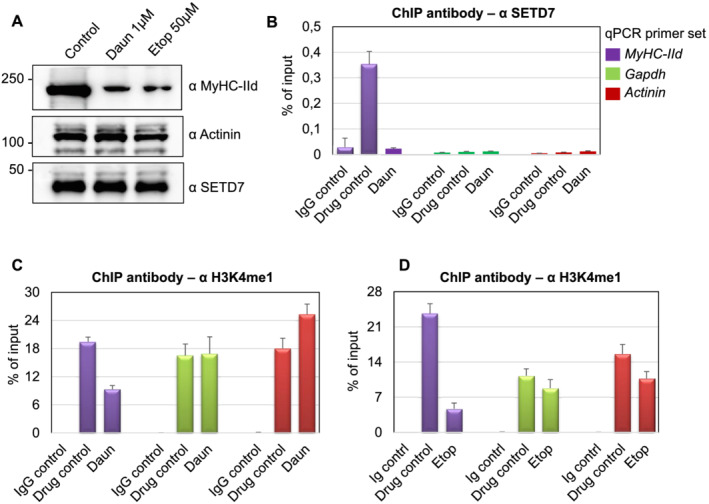
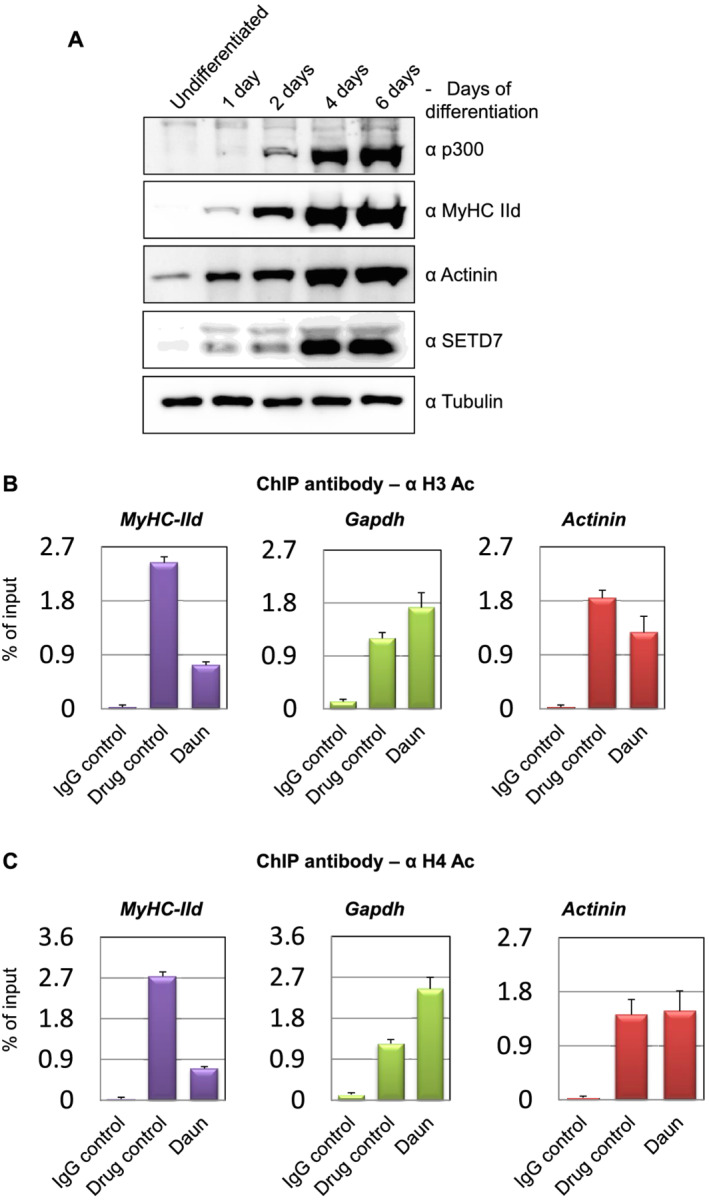
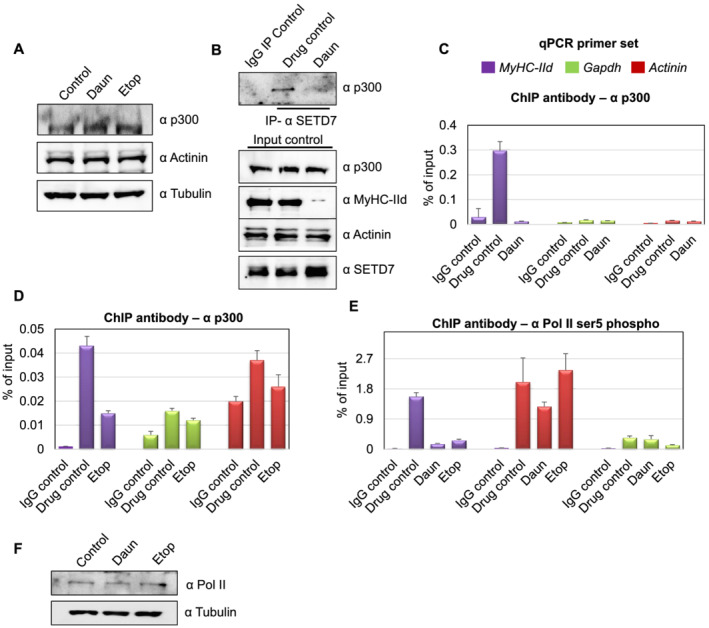
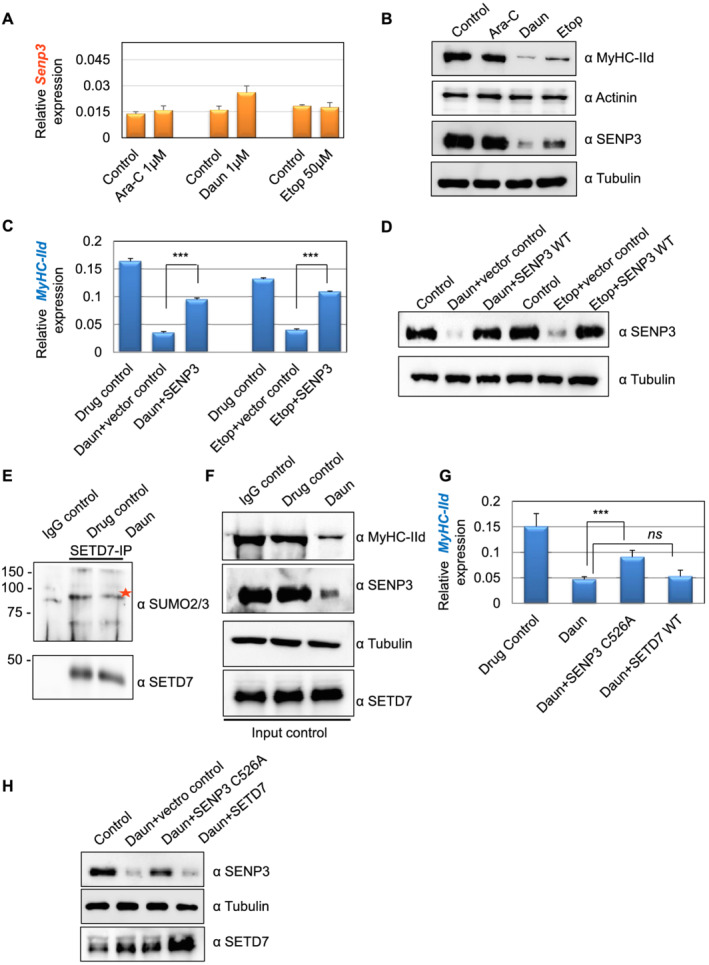
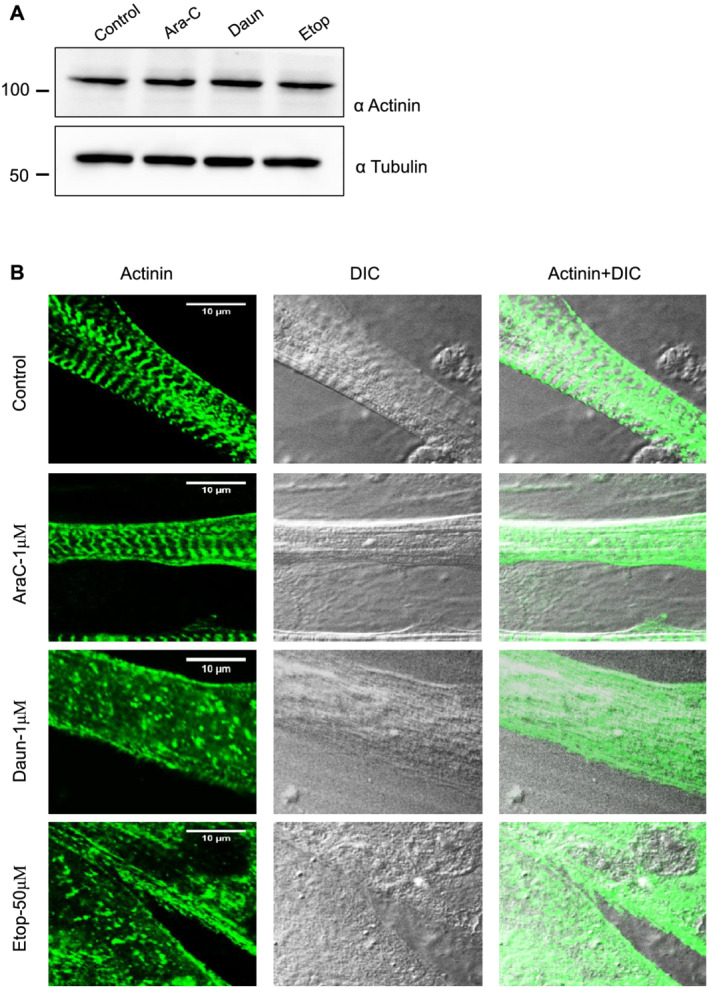
Here, we demonstrate that chemotherapeutic drugs disrupt sarcomere organization and thereby the contractile ability of skeletal muscle cells. The sarcomere disorganization results from severe loss of molecular motor protein MyHC-II upon drug treatment. We identified that drugs impede chromatin targeting of SETD7 histone methyltransferase and disrupt association and synergetic function of SETD7 with p300 histone acetyltransferase. The compromised transcriptional activity of histone methyltransferase and acetyltransferase causes reduced histone acetylation and low occupancy of active RNA polymerase II on MyHC-II, promoting drastic down-regulation of MyHC-II expression (~3.6-fold and ~4.5-fold reduction of MyHC-IId mRNA levels in Daun and Etop treatment, respectively. P < 0.0001). For MyHC-IIa, gene expression was down-regulated by ~2.6-fold and ~4.5-fold in Daun and Etop treatment, respectively (P < 0.0001). Very interestingly, the drugs destabilize SUMO deconjugase SENP3. Reduction in SENP3 protein level leads to deregulation of SETD7-p300 function. Importantly, we identified that SUMO deconjugation independent role of SENP3 regulates SETD7-p300 functional axis.
The results show that the drugs critically alter SENP3-dependent synergistic action of histone-modifying enzymes in muscle cells. Collectively, we defined a unique epigenetic mechanism targeted by distinct chemotherapeutic drugs, triggering chemotherapy-induced cachexia.
化疗是癌症患者的一线治疗方法。然而,其副作用会导致严重的肌肉萎缩或化疗诱导的恶病质。此前,已证明NF-κB/MuRF1依赖性途径可诱导化疗诱导的恶病质。我们推测化疗对肌肉的急性附带毒性作用可能涉及其他未知途径,从而促进化疗诱导的肌肉萎缩。在本研究中,我们研究了化疗药物的不同作用,并探究是否有其他分子机制导致恶病质。
我们使用小鼠卫星干细胞来源的原代肌肉细胞和小鼠C2C12祖细胞来源的分化肌管作为模型系统来测试药物的作用。测试了广泛使用的化疗药物,如柔红霉素(Daun)、依托泊苷(Etop)和阿糖胞苷(Ara-C)。通过染色质免疫沉淀、内源性基因表达谱分析、免疫共沉淀、互补分析和共聚焦显微镜等方法,检测药物在表观遗传、转录和蛋白质水平上影响肌肉细胞组织的分子机制。通过电刺激肌管来监测药物处理后肌管的收缩能力(兴奋-收缩偶联),以此检测肌管功能。
在此,我们证明化疗药物会破坏肌节组织,进而破坏骨骼肌细胞的收缩能力。药物处理后,肌节紊乱是由于分子运动蛋白MyHC-II严重缺失所致。我们发现药物会阻碍SETD7组蛋白甲基转移酶的染色质靶向作用,并破坏SETD7与p300组蛋白乙酰转移酶的结合及协同功能。组蛋白甲基转移酶和乙酰转移酶受损的转录活性导致组蛋白乙酰化减少,活性RNA聚合酶II在MyHC-II上的占有率降低,从而促进MyHC-II表达的急剧下调(柔红霉素和依托泊苷处理后,MyHC-IId mRNA水平分别降低约3.6倍和约4.5倍。P < 0.0001)。对于MyHC-IIa,在柔红霉素和依托泊苷处理中,基因表达分别下调约2.6倍和约4.5倍(P < 0.0001)。非常有趣的是,这些药物会使SUMO去共轭酶SENP3不稳定。SENP3蛋白水平的降低导致SETD7-p300功能失调。重要的是,我们发现SENP3的SUMO去共轭独立作用调节SETD7-p300功能轴。
结果表明,这些药物严重改变了肌肉细胞中SENP3依赖性组蛋白修饰酶的协同作用。总体而言,我们定义了一种独特的表观遗传机制,该机制是不同化疗药物的作用靶点,可引发化疗诱导的恶病质。