Leibniz Institute for Farm Animal Biology (FBN), Institute of Genetics and Biometry, Wilhelm-Stahl-Allee 2, 18196, Dummerstorf, Germany.
Genet Sel Evol. 2020 Dec 14;52(1):73. doi: 10.1186/s12711-020-00593-z.
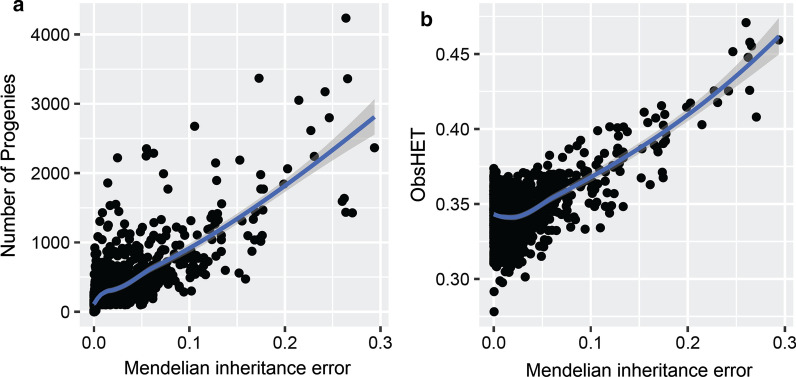
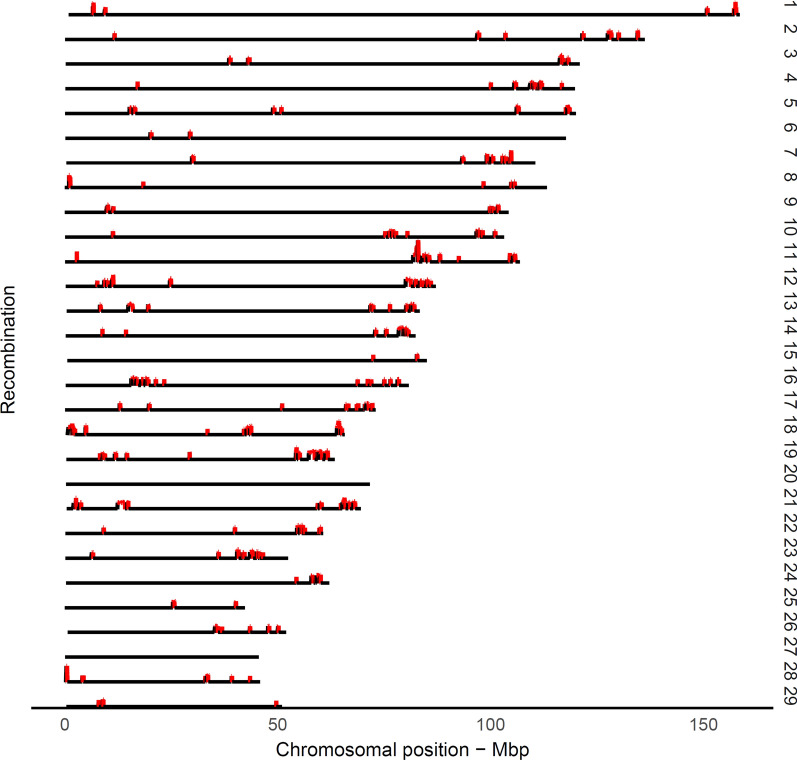
Recombination is a process by which chromosomes are broken and recombine to generate new combinations of alleles, therefore playing a major role in shaping genome variation. Recombination frequencies ([Formula: see text]) between markers are used to construct genetic maps, which have important implications in genomic studies. Here, we report a recombination map for 44,696 autosomal single nucleotide polymorphisms (SNPs) according to the coordinates of the most recent bovine reference assembly. The recombination frequencies were estimated across 876 half-sib families with a minimum number of 39 and maximum number of 4236 progeny, comprising over 367 K genotyped German Holstein animals.
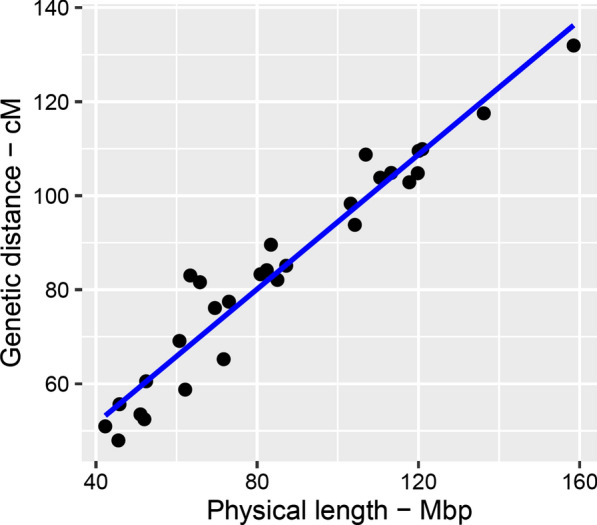
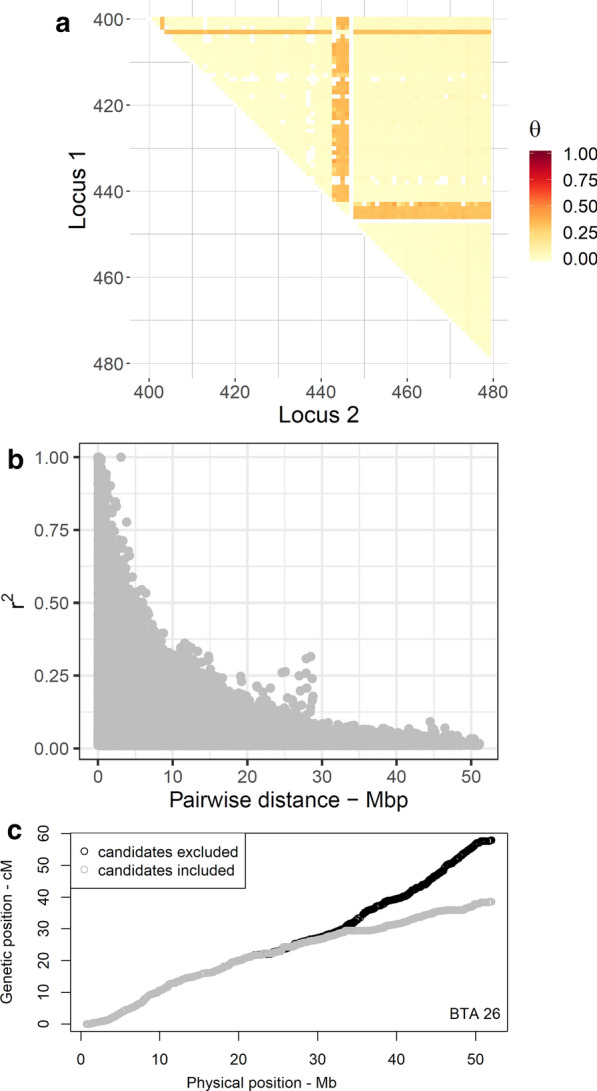
Genome-wide, over 8.9 million paternal recombination events were identified by investigating adjacent markers. The recombination map spans 24.43 Morgan (M) for a chromosomal length of 2486 Mbp and an average of ~ 0.98 cM/Mbp, which concords with the available pedigree-based linkage maps. Furthermore, we identified 971 putative recombination hotspot intervals (defined as [Formula: see text] > 2.5 standard deviations greater than the mean). The hotspot regions were non-uniformly distributed as sharp and narrow peaks, corresponding to ~ 5.8% of the recombination that has taken place in only ~ 2.4% of the genome. We verified genetic map length by applying a likelihood-based approach for the estimation of recombination rate between all intra-chromosomal marker pairs. This resulted in a longer autosomal genetic length for male cattle (25.35 cM) and in the localization of 51 putatively misplaced SNPs in the genome assembly.
Given the fact that this map is built on the coordinates of the ARS-UCD1.2 assembly, our results provide the most updated genetic map yet available for the cattle genome.
重组是一个染色体断裂和重新组合以产生等位基因新组合的过程,因此在塑造基因组变异方面起着重要作用。标记之间的重组频率([Formula: see text])用于构建遗传图谱,这对基因组研究具有重要意义。在这里,我们根据最新的牛参考组装坐标报告了 44696 个常染色体单核苷酸多态性(SNP)的重组图谱。在 876 个半同胞家系中估计了重组频率,这些家系的最小后代数为 39 个,最大后代数为 4236 个,包含超过 36.7 万头经过基因分型的德国荷斯坦牛。
在研究相邻标记时,在全基因组范围内共鉴定出超过 890 万次父系重组事件。重组图谱跨越 24.43 摩根(M),染色体长度为 2486 Mbp,平均每 0.98 cM/Mbp,与现有基于系谱的连锁图谱一致。此外,我们还鉴定出 971 个可能的重组热点区间(定义为[Formula: see text]>2.5 个标准差大于平均值)。热点区域的分布不均匀,呈尖锐而狭窄的峰值,对应于仅在基因组的 2.4%中发生的重组的约 5.8%。我们通过应用基于似然的方法来估计所有染色体内标记对之间的重组率来验证遗传图谱的长度。这导致雄性牛的常染色体遗传长度更长(25.35 cM),并在基因组组装中定位了 51 个可能错位的 SNP。
鉴于该图谱是基于 ARS-UCD1.2 组装的坐标构建的,我们的结果为牛基因组提供了迄今为止最新的遗传图谱。