Unit of Animal Genomics, GIGA-R and Faculty of Veterinary Medicine, University of Liège (B34), 4000, Liège, Belgium.
Animal Genomics, ETH Zürich, 8092, Zürich, Switzerland.
BMC Genomics. 2022 Feb 15;23(1):130. doi: 10.1186/s12864-022-08354-6.
Accurate haplotype reconstruction is required in many applications in quantitative and population genomics. Different phasing methods are available but their accuracy must be evaluated for samples with different properties (population structure, marker density, etc.). We herein took advantage of whole-genome sequence data available for a Holstein cattle pedigree containing 264 individuals, including 98 trios, to evaluate several population-based phasing methods. This data represents a typical example of a livestock population, with low effective population size, high levels of relatedness and long-range linkage disequilibrium.
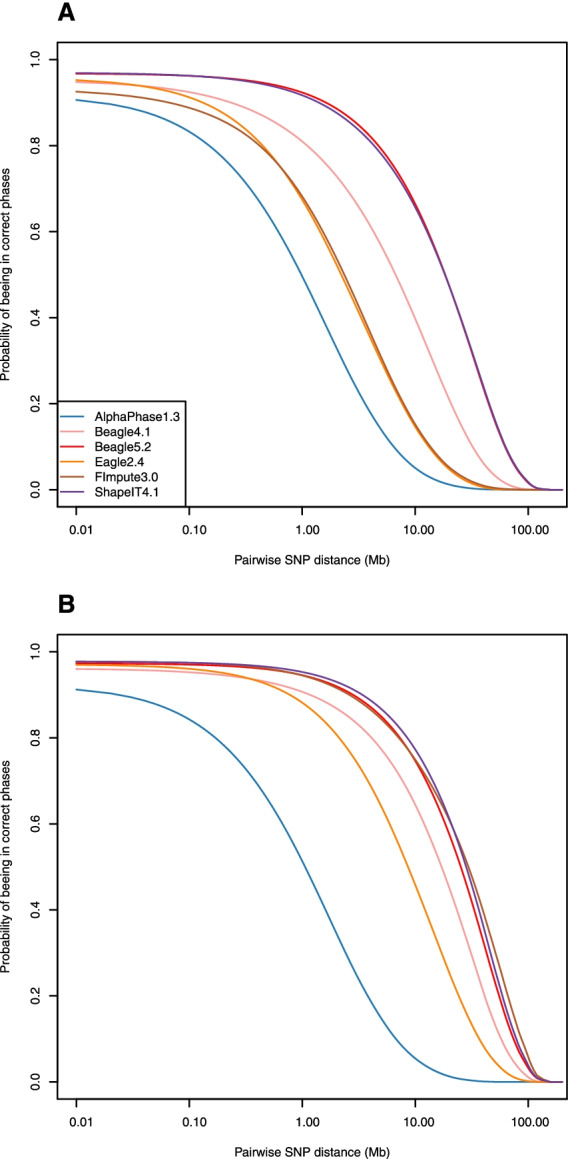
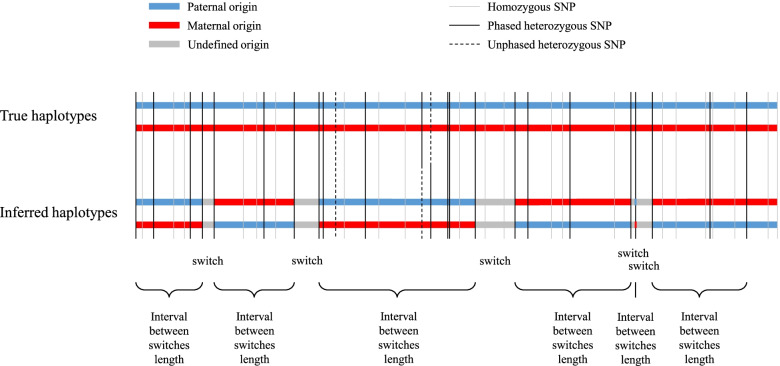
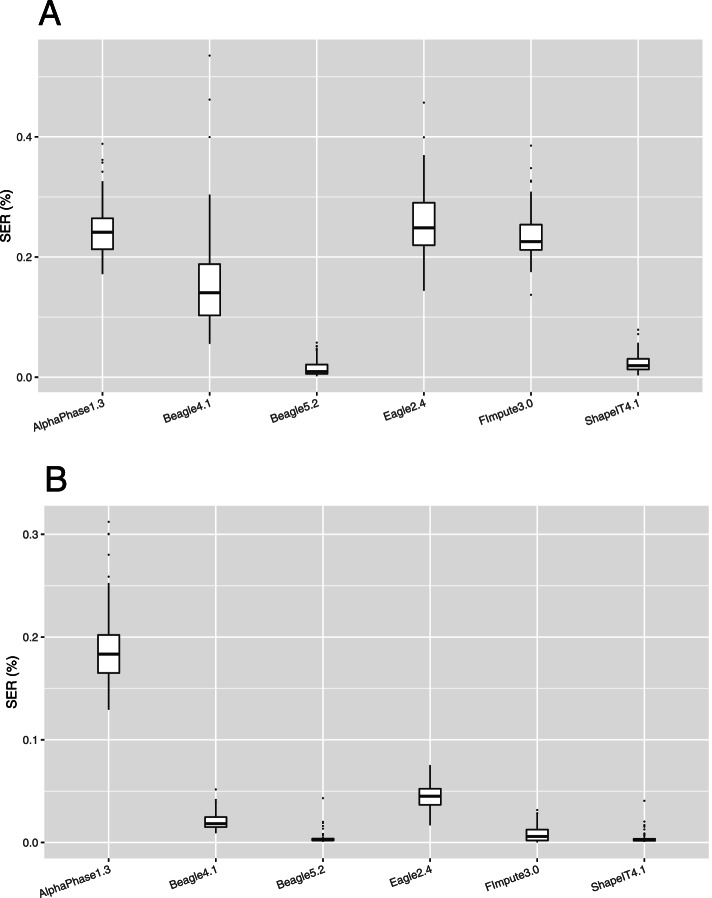
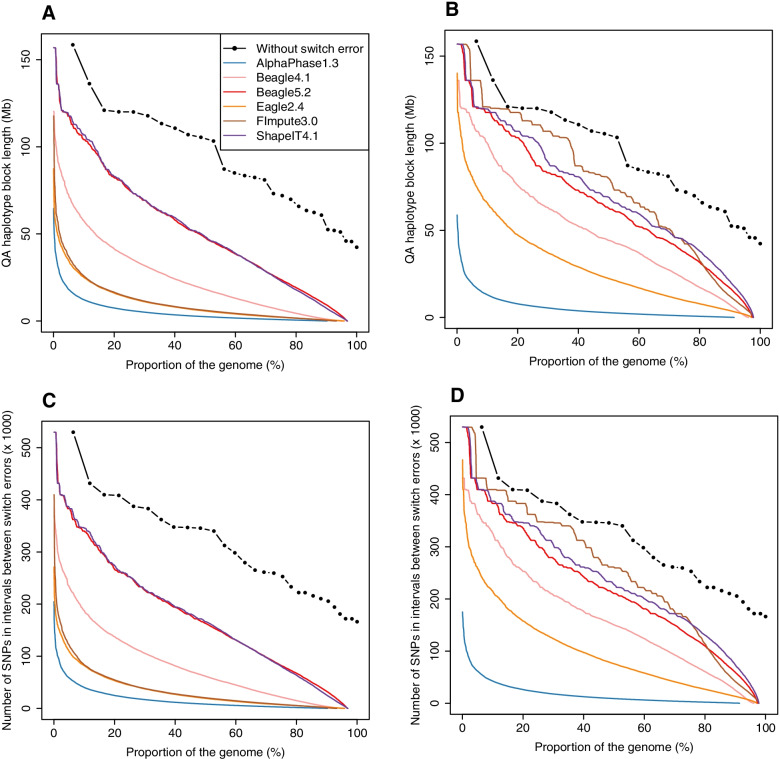
After stringent filtering of our sequence data, we evaluated several population-based phasing programs including one or more versions of AlphaPhase, ShapeIT, Beagle, Eagle and FImpute. To that end we used 98 individuals having both parents sequenced for validation. Their haplotypes reconstructed based on Mendelian segregation rules were considered the gold standard to assess the performance of population-based methods in two scenarios. In the first one, only these 98 individuals were phased, while in the second one, all the 264 sequenced individuals were phased simultaneously, ignoring the pedigree relationships. We assessed phasing accuracy based on switch error counts (SEC) and rates (SER), lengths of correctly phased haplotypes and the probability that there is no phasing error between a pair of SNPs as a function of their distance. For most evaluated metrics or scenarios, the best software was either ShapeIT4.1 or Beagle5.2, both methods resulting in particularly high phasing accuracies. For instance, ShapeIT4.1 achieved a median SEC of 50 per individual and a mean haplotype block length of 24.1 Mb (scenario 2). These statistics are remarkable since the methods were evaluated with a map of 8,400,000 SNPs, and this corresponds to only one switch error every 40,000 phased informative markers. When more relatives were included in the data (scenario 2), FImpute3.0 reconstructed extremely long segments without errors.
We report extremely high phasing accuracies in a typical livestock sample. ShapeIT4.1 and Beagle5.2 proved to be the most accurate, particularly for phasing long segments and in the first scenario. Nevertheless, most tools achieved high accuracy at short distances and would be suitable for applications requiring only local haplotypes.
在定量和群体基因组学的许多应用中,需要准确的单倍型重建。有不同的相位方法,但必须针对具有不同属性(群体结构、标记密度等)的样本评估其准确性。我们在此利用了一个荷斯坦牛系谱的全基因组序列数据,该系谱包含 264 个个体,包括 98 个三亲家庭,用于评估几种基于群体的相位方法。该数据代表了一个典型的家畜群体,有效群体规模小,亲缘关系高,长程连锁不平衡。
在对我们的序列数据进行严格过滤后,我们评估了几种基于群体的相位程序,包括一个或多个版本的 AlphaPhase、ShapeIT、Beagle、Eagle 和 FImpute。为此,我们使用了 98 个具有双亲测序的个体进行验证。根据孟德尔分离规则重建的单倍型被认为是评估基于群体的方法在两种情况下表现的金标准。在第一种情况下,只有这 98 个个体被相位化,而在第二种情况下,264 个测序的个体同时被相位化,忽略了系谱关系。我们根据转换错误计数(SEC)和速率(SER)、正确相位化单倍型的长度以及一对 SNP 之间是否存在相位错误的概率来评估相位精度,作为其距离的函数。对于大多数评估指标或场景,最好的软件是 ShapeIT4.1 或 Beagle5.2,这两种方法都产生了特别高的相位精度。例如,ShapeIT4.1 每个个体的中位数 SEC 为 50,平均单倍型块长度为 24.1 Mb(场景 2)。这些统计数据非常显著,因为方法是在一个包含 840 万个 SNP 的图谱上进行评估的,这相当于每 40,000 个相位化的信息标记只有一个转换错误。当更多的亲属被纳入数据(场景 2)时,FImpute3.0 重建了没有错误的极其长的片段。
我们在一个典型的家畜样本中报告了极高的相位精度。ShapeIT4.1 和 Beagle5.2 被证明是最准确的,特别是对于长片段的相位化和第一种情况。然而,大多数工具在短距离上都达到了很高的准确性,并且适用于仅需要局部单倍型的应用。