Müller Julia, Timmermann Aline, Henning Lukas, Müller Hendrik, Steinhäuser Christian, Bedner Peter
Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, Bonn, Germany.
Front Neurol. 2020 Dec 18;11:614923. doi: 10.3389/fneur.2020.614923. eCollection 2020.
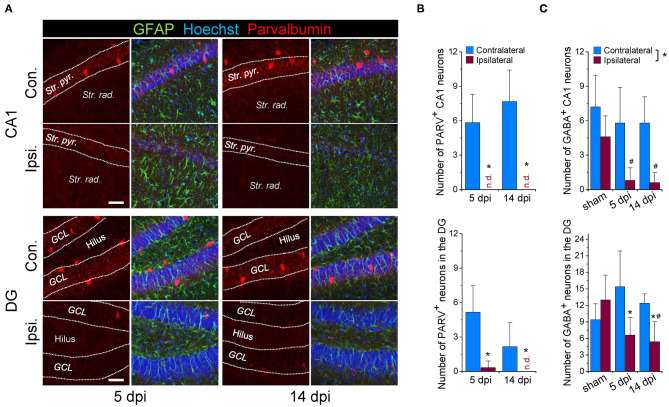
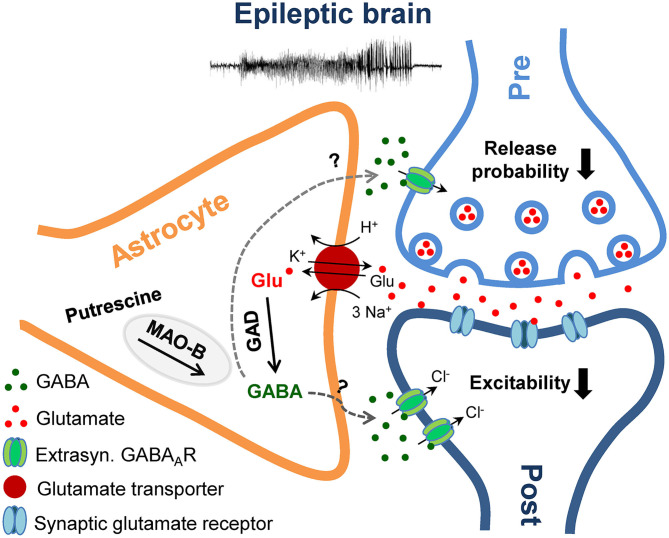
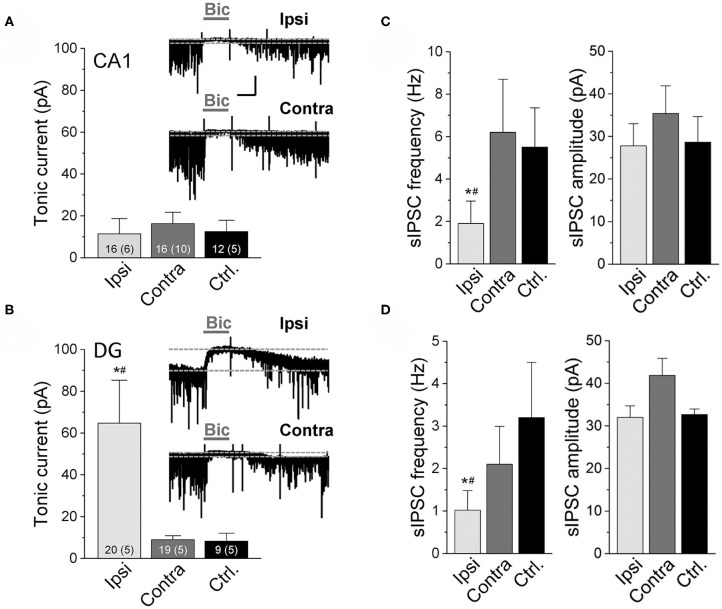
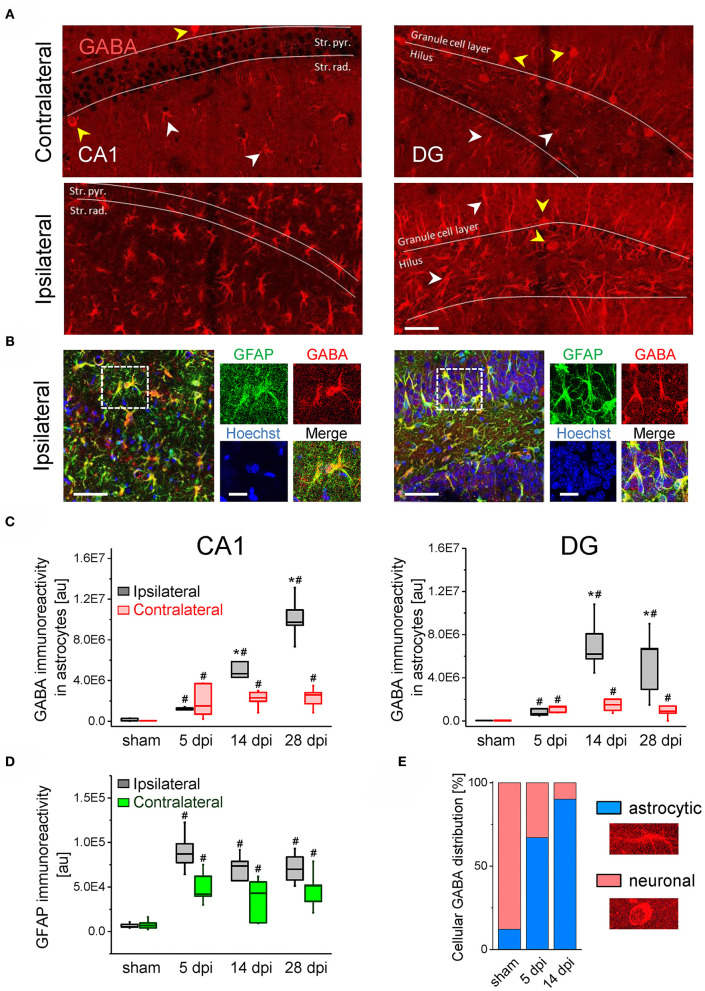
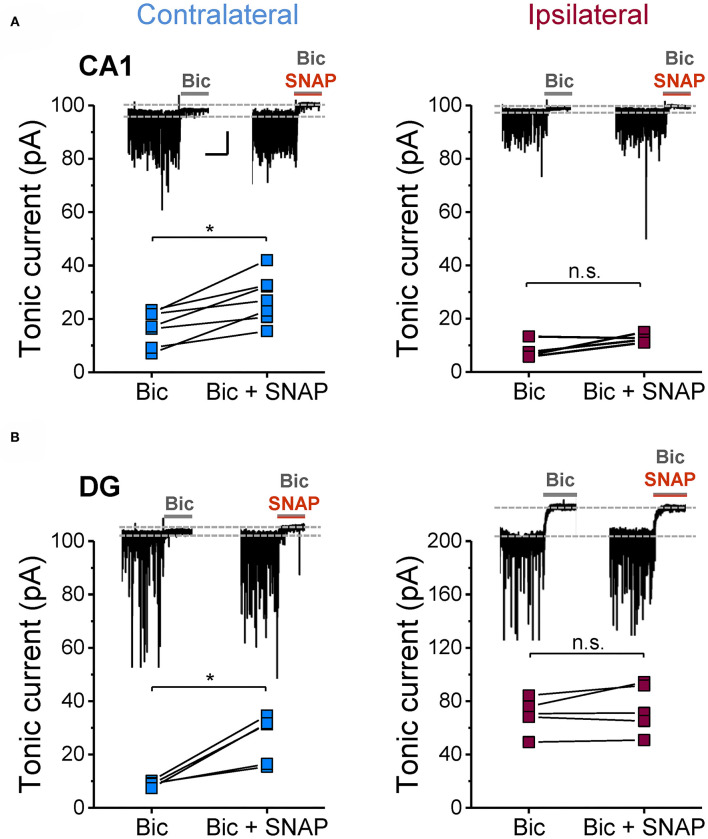
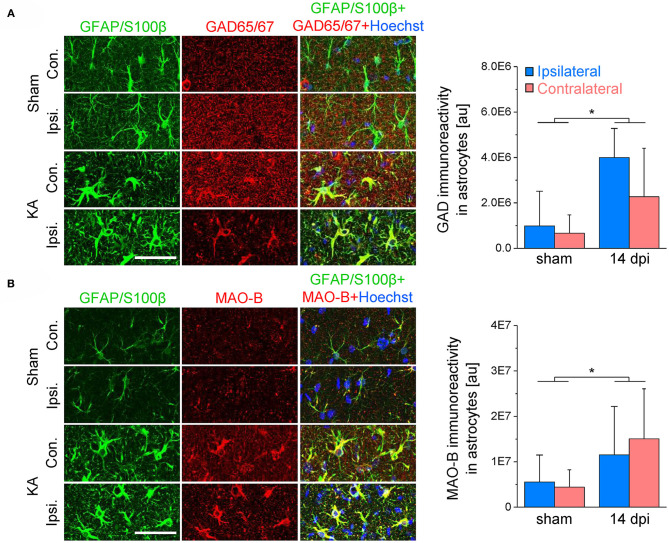
An imbalance of excitation and inhibition has been associated with the pathophysiology of epilepsy. Loss of GABAergic interneurons and/or synaptic inhibition has been shown in various epilepsy models and in human epilepsy. Despite this loss, several studies reported preserved or increased tonic GABA receptor-mediated currents in epilepsy, raising the question of the source of the inhibitory transmitter. We used the unilateral intracortical kainate mouse model of temporal lobe epilepsy (TLE) with hippocampal sclerosis (HS) to answer this question. In our model we observed profound loss of interneurons in the sclerotic hippocampal CA1 region and dentate gyrus already 5 days after epilepsy induction. Consistent with the literature, the absence of interneurons caused no reduction of tonic inhibition of CA1 pyramidal neurons. In dentate granule cells the inhibitory currents were even increased in epileptic tissue. Intriguingly, immunostaining of brain sections from epileptic mice with antibodies against GABA revealed strong and progressive accumulation of the neurotransmitter in reactive astrocytes. Pharmacological inhibition of the astrocytic GABA transporter GAT3 did not affect tonic inhibition in the sclerotic hippocampus, suggesting that this transporter is not responsible for astrocytic GABA accumulation or release. Immunostaining further indicated that both decarboxylation of glutamate and putrescine degradation accounted for the increased GABA levels in reactive astrocytes. Together, our data provide evidence that the preserved tonic inhibitory currents in the epileptic brain are mediated by GABA overproduction and release from astrocytes. A deeper understanding of the underlying mechanisms may lead to new strategies for antiepileptic drug therapy.
兴奋与抑制的失衡与癫痫的病理生理学相关。在各种癫痫模型和人类癫痫中均已显示γ-氨基丁酸(GABA)能中间神经元的丧失和/或突触抑制作用。尽管存在这种丧失,但多项研究报告称癫痫中强直GABA受体介导的电流保持不变或增加,这就引发了抑制性递质来源的问题。我们使用伴有海马硬化(HS)的颞叶癫痫(TLE)单侧皮质内注射红藻氨酸盐小鼠模型来回答这个问题。在我们的模型中,癫痫诱导后仅5天,我们就观察到硬化海马CA1区和齿状回中的中间神经元大量丧失。与文献一致,中间神经元的缺失并未导致CA1锥体神经元的强直抑制作用减弱。在癫痫组织中,齿状颗粒细胞的抑制性电流甚至增加。有趣的是,用抗GABA抗体对癫痫小鼠脑切片进行免疫染色显示,神经递质在反应性星形胶质细胞中强烈且逐渐积累。对星形胶质细胞GABA转运体GAT3进行药理学抑制并不影响硬化海马中的强直抑制作用,这表明该转运体与星形胶质细胞GABA的积累或释放无关。免疫染色进一步表明,谷氨酸脱羧和腐胺降解均导致反应性星形胶质细胞中GABA水平升高。总之,我们的数据提供了证据,表明癫痫脑中保留的强直抑制电流是由星形胶质细胞过量产生和释放GABA介导的。对其潜在机制的更深入理解可能会带来抗癫痫药物治疗的新策略。