Departments of Pathology.
Oncology, The Johns Hopkins Medical Institutions, Baltimore, MD.
Am J Surg Pathol. 2021 May 1;45(5):653-661. doi: 10.1097/PAS.0000000000001656.
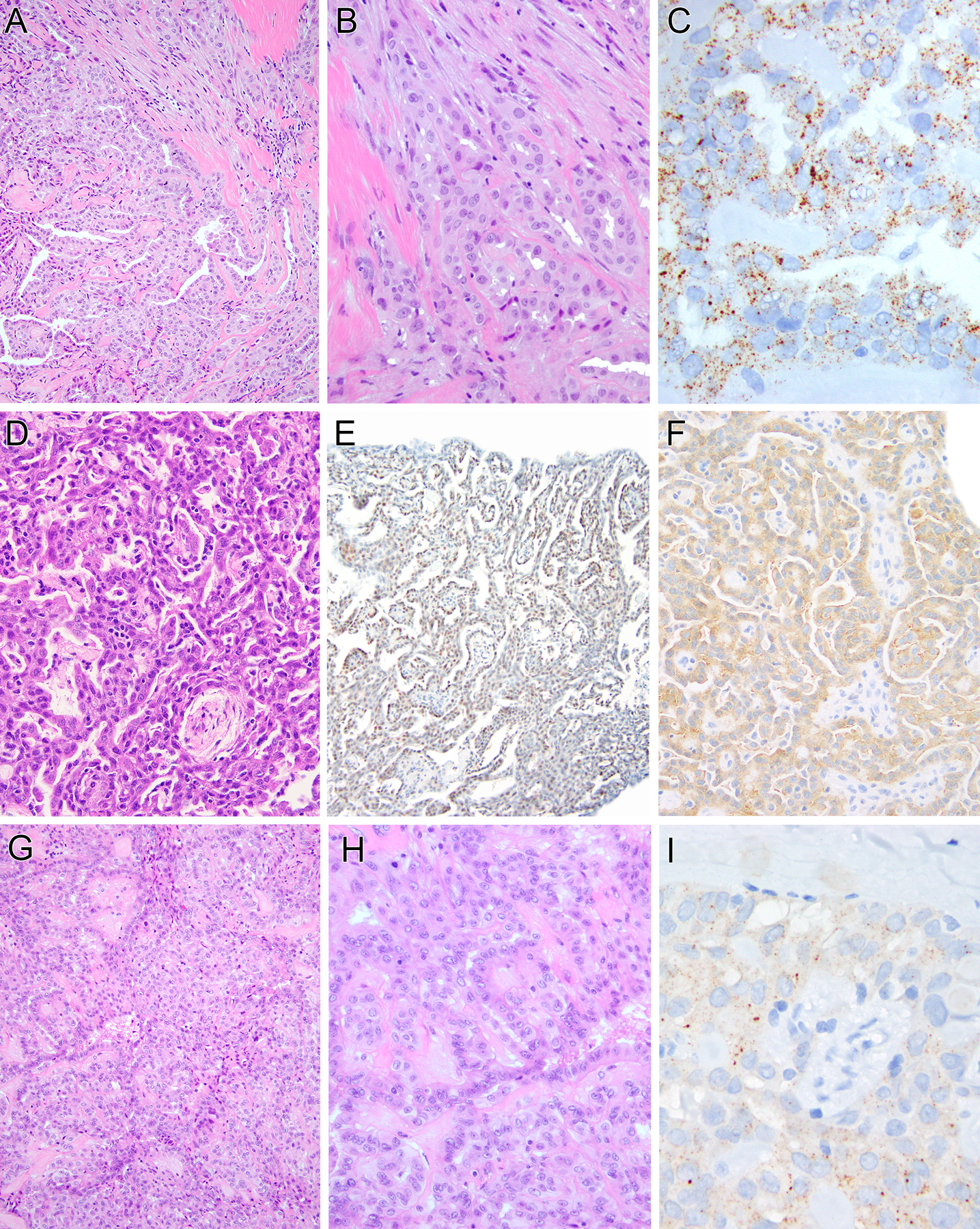
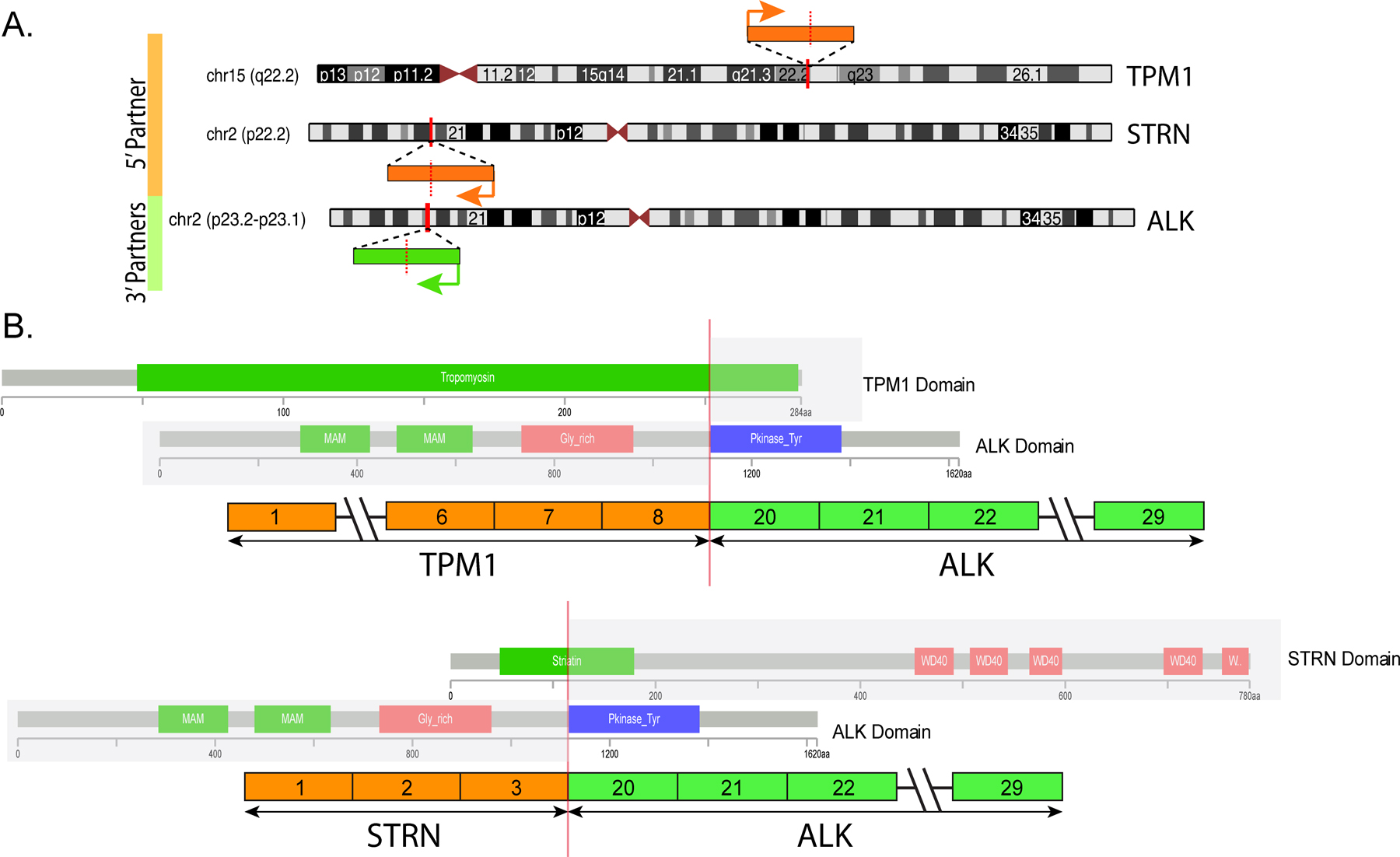
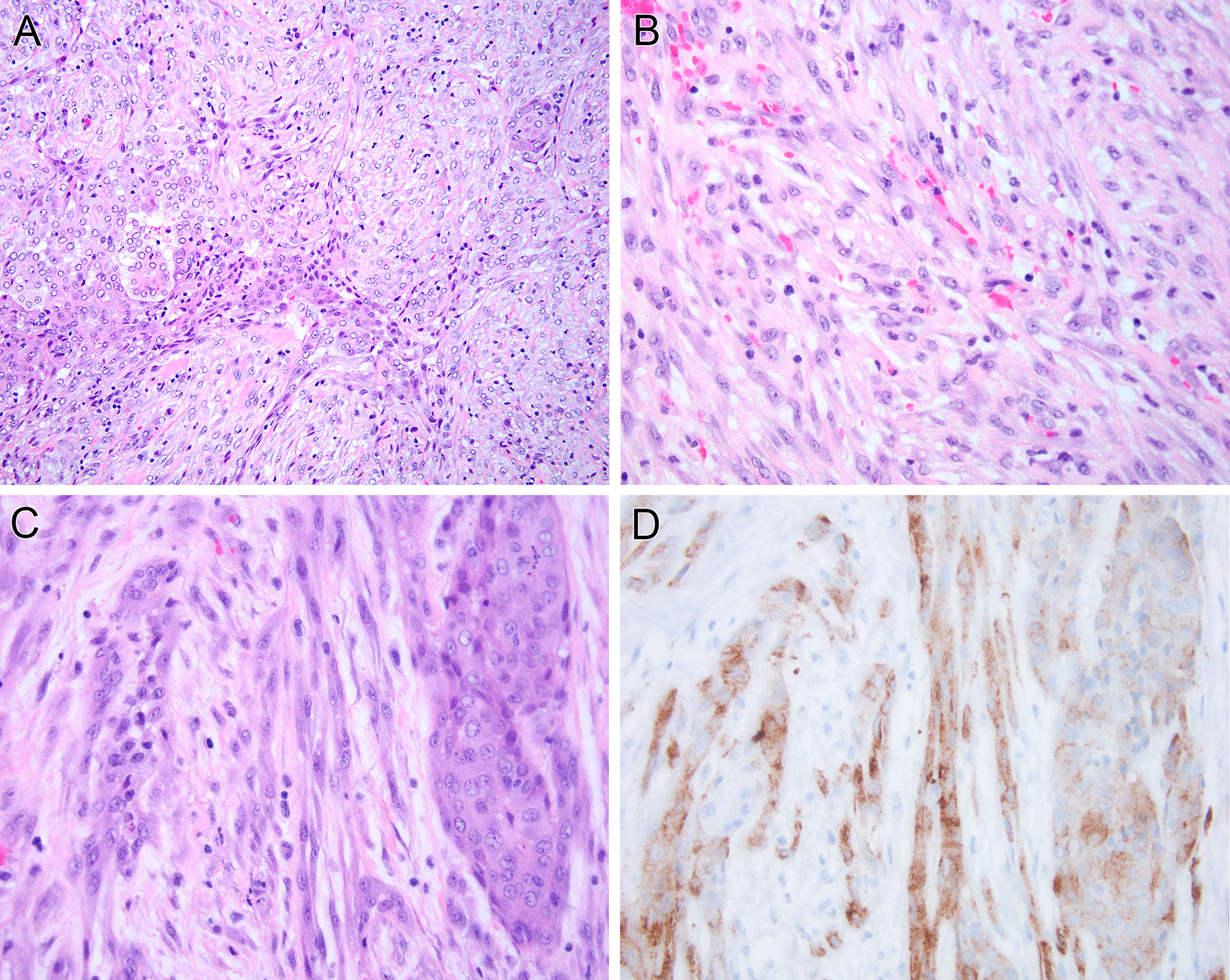
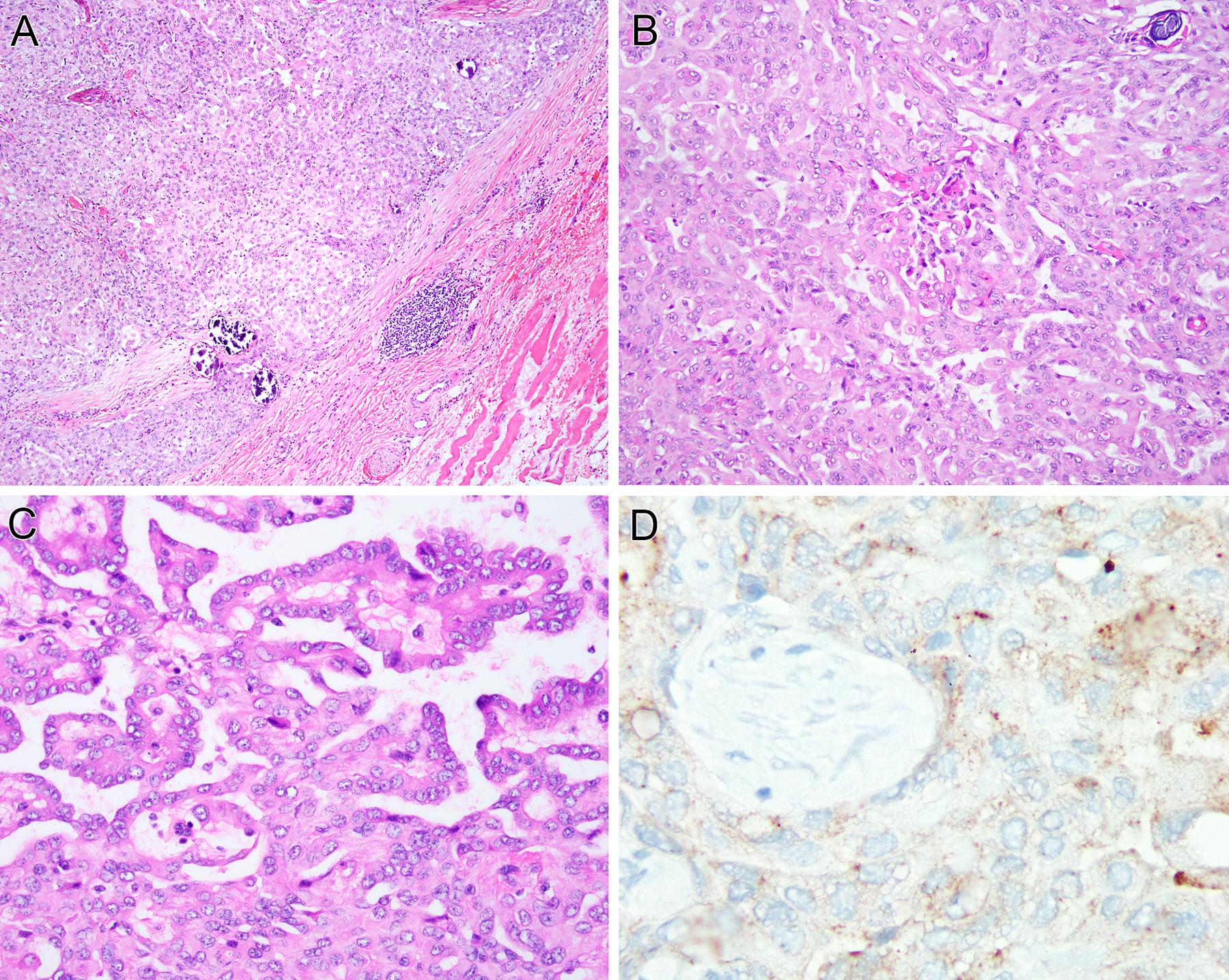
Pediatric mesotheliomas are rare and their pathogenesis remains undefined. In this study, we report 5 cases of malignant mesothelioma in children, characterized by fusions involving the anaplastic lymphoma kinase (ALK) gene. Four cases occurred in females involving the abdominal cavity and were characterized by a pure epithelioid morphology. The fifth arose in the tunica vaginalis of a 15-year-old male and displayed a biphasic epithelioid-sarcomatoid phenotype. All cases demonstrated the classic morphologic and immunohistochemical features of malignant mesothelioma, including tubulopapillary architecture and cuboidal epithelioid cells with eosinophilic cytoplasm and uniform nuclei with vesicular chromatin. Immunohistochemically, all cases showed labeling for ALK, cytokeratins, WT1, and calretinin, while lacking expression of adenocarcinoma immunomarkers. Four cases demonstrated weak-moderate labeling for PAX8 protein, which resulted in diagnostic challenges with primary peritoneal serous carcinoma. The ALK genetic abnormalities were investigated by a combination of molecular methods. Archer FusionPlex was performed in 2 cases, showing fusions between ALK with either STRN or TPM1 genes, resulting in a transcript that retained the ALK kinase domain. One case was further studied by DNA targeted sequencing, but no additional genetic alterations were observed. In 1 case, cytogenetic analysis showed the presence of a t(2;15)(p23;q22) and fluorescence in situ hybridization confirmed the ALK gene break-apart. In the remaining 2 cases, ALK gene rearrangements were demonstrated by fluorescence in situ hybridization. Unlike adult mesotheliomas, which are tightly linked to asbestos exposure, often show loss of BAP1 expression and have complex karyotypes, ALK-rearranged mesothelioma appears to be similar to other fusion-positive mesotheliomas, such as those harboring EWSR1/FUS-ATF1 fusions, sharing significant morphologic overlap, occurring in young patients and displaying a simple, translocation-driven genetic profile.
儿科间皮瘤罕见,其发病机制尚不清楚。在本研究中,我们报告了 5 例儿童恶性间皮瘤病例,这些病例均存在涉及间变性淋巴瘤激酶(ALK)基因的融合。4 例发生于女性,位于腹腔,具有纯上皮样形态。第 5 例发生于 15 岁男性的鞘膜,表现为上皮样-肉瘤样双相表型。所有病例均表现出恶性间皮瘤的典型形态学和免疫组织化学特征,包括小管状乳头状结构和嗜酸性细胞质的立方上皮样细胞,核均匀,染色质呈泡状。免疫组化染色显示所有病例均表达 ALK、细胞角蛋白、WT1 和钙视网膜蛋白,而缺乏腺癌免疫标志物的表达。4 例病例显示 PAX8 蛋白弱阳性-中度表达,这导致与原发性腹膜浆液性癌的诊断存在挑战。通过结合分子方法研究 ALK 基因异常。在 2 例病例中进行了 Archer FusionPlex,显示 ALK 与 STRN 或 TPM1 基因融合,导致保留 ALK 激酶结构域的转录本。1 例病例进一步进行了 DNA 靶向测序,但未观察到其他遗传改变。在 1 例病例中,细胞遗传学分析显示存在 t(2;15)(p23;q22),荧光原位杂交证实 ALK 基因断裂。在其余 2 例病例中,通过荧光原位杂交显示存在 ALK 基因重排。与紧密关联于石棉暴露的成人间皮瘤不同,儿童间皮瘤通常表现为 BAP1 表达缺失和复杂的核型,ALK 重排型间皮瘤似乎与其他融合阳性间皮瘤相似,如具有 EWSR1/FUS-ATF1 融合的间皮瘤,具有显著的形态重叠,发生于年轻患者,具有简单的、易位驱动的遗传特征。