Department of Microbiology, Immunology and Genetics, University of North Texas Health Science Center, 3500 Camp Bowie Blvd, Fort Worth, TX, 76107, USA.
BMC Bioinformatics. 2021 Jan 6;22(1):12. doi: 10.1186/s12859-020-03945-0.
Multi-locus genotype data are widely used in population genetics and disease studies. In evaluating the utility of multi-locus data, the independence of markers is commonly considered in many genomic assessments. Generally, pairwise non-random associations are tested by linkage disequilibrium; however, the dependence of one panel might be triplet, quartet, or other. Therefore, a compatible and user-friendly software is necessary for testing and assessing the global linkage disequilibrium among mixed genetic data.
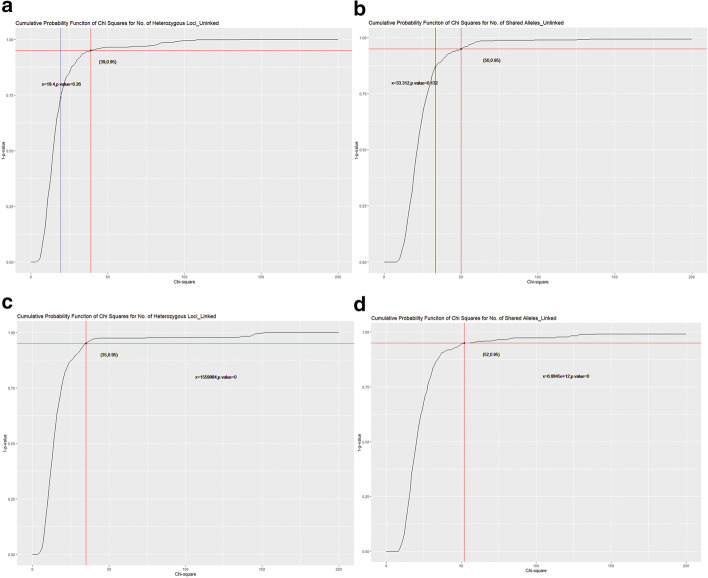
This study describes a software package for testing the mutual independence of mixed genetic datasets. Mutual independence is defined as no non-random associations among all subsets of the tested panel. The new R package "mixIndependR" calculates basic genetic parameters like allele frequency, genotype frequency, heterozygosity, Hardy-Weinberg equilibrium, and linkage disequilibrium (LD) by mutual independence from population data, regardless of the type of markers, such as simple nucleotide polymorphisms, short tandem repeats, insertions and deletions, and any other genetic markers. A novel method of assessing the dependence of mixed genetic panels is developed in this study and functionally analyzed in the software package. By comparing the observed distribution of two common summary statistics (the number of heterozygous loci [K] and the number of share alleles [X]) with their expected distributions under the assumption of mutual independence, the overall independence is tested.
The package "mixIndependR" is compatible to all categories of genetic markers and detects the overall non-random associations. Compared to pairwise disequilibrium, the approach described herein tends to have higher power, especially when number of markers is large. With this package, more multi-functional or stronger genetic panels can be developed, like mixed panels with different kinds of markers. In population genetics, the package "mixIndependR" makes it possible to discover more about admixture of populations, natural selection, genetic drift, and population demographics, as a more powerful method of detecting LD. Moreover, this new approach can optimize variants selection in disease studies and contribute to panel combination for treatments in multimorbidity. Application of this approach in real data is expected in the future, and this might bring a leap in the field of genetic technology.
The R package mixIndependR, is available on the Comprehensive R Archive Network (CRAN) at: https://cran.r-project.org/web/packages/mixIndependR/index.html .
多位点基因型数据广泛应用于群体遗传学和疾病研究。在评估多位点数据的效用时,许多基因组评估通常考虑标记的独立性。通常,通过连锁不平衡来测试标记之间的非随机关联;然而,一个面板的依赖性可能是三联体、四重体或其他形式。因此,需要一个兼容且用户友好的软件来测试和评估混合遗传数据的全局连锁不平衡。
本研究描述了一个用于测试混合遗传数据集相互独立性的软件包。相互独立性定义为测试面板的所有子集之间没有非随机关联。新的 R 包“mixIndependR”通过相互独立性从群体数据中计算基本遗传参数,如等位基因频率、基因型频率、杂合度、哈迪-温伯格平衡和连锁不平衡(LD),而不管标记的类型如何,如简单核苷酸多态性、短串联重复、插入和缺失以及任何其他遗传标记。本研究中开发了一种评估混合遗传面板依赖性的新方法,并在软件包中进行了功能分析。通过比较两个常见汇总统计量(杂合基因座数[K]和共享等位基因数[X])的观测分布与相互独立性假设下的预期分布,检验整体独立性。
该软件包“mixIndependR”与所有类别的遗传标记兼容,并检测整体非随机关联。与成对不平衡相比,本文描述的方法具有更高的功效,特别是当标记数量较大时。使用此软件包,可以开发更多多功能或更强的遗传面板,例如具有不同类型标记的混合面板。在群体遗传学中,该软件包“mixIndependR”使得发现更多关于群体混合、自然选择、遗传漂变和群体人口统计学的信息成为可能,作为检测 LD 的更强大方法。此外,这种新方法可以优化疾病研究中的变体选择,并有助于多疾病治疗中的面板组合。预计未来将在实际数据中应用这种方法,这可能会推动遗传技术领域的发展。
R 包 mixIndependR 可在 Comprehensive R Archive Network (CRAN) 上获得,网址为:https://cran.r-project.org/web/packages/mixIndependR/index.html。