Nilo-Poyanco Ricardo, Moraga Carol, Benedetto Gianfranco, Orellana Ariel, Almeida Andrea Miyasaka
Escuela de Biotecnología, Facultad de Ciencias, Universidad Mayor, Camino La Pirámide, 5750, Huechuraba, Chile.
Université Claude Bernard Lyon 1, 69622, Villeurbanne, France.
BMC Genomics. 2021 Jan 6;22(1):17. doi: 10.1186/s12864-020-07299-y.
Fruit ripening in Prunus persica melting varieties involves several physiological changes that have a direct impact on the fruit organoleptic quality and storage potential. By studying the proteomic differences between the mesocarp of mature and ripe fruit, it would be possible to highlight critical molecular processes involved in the fruit ripening.
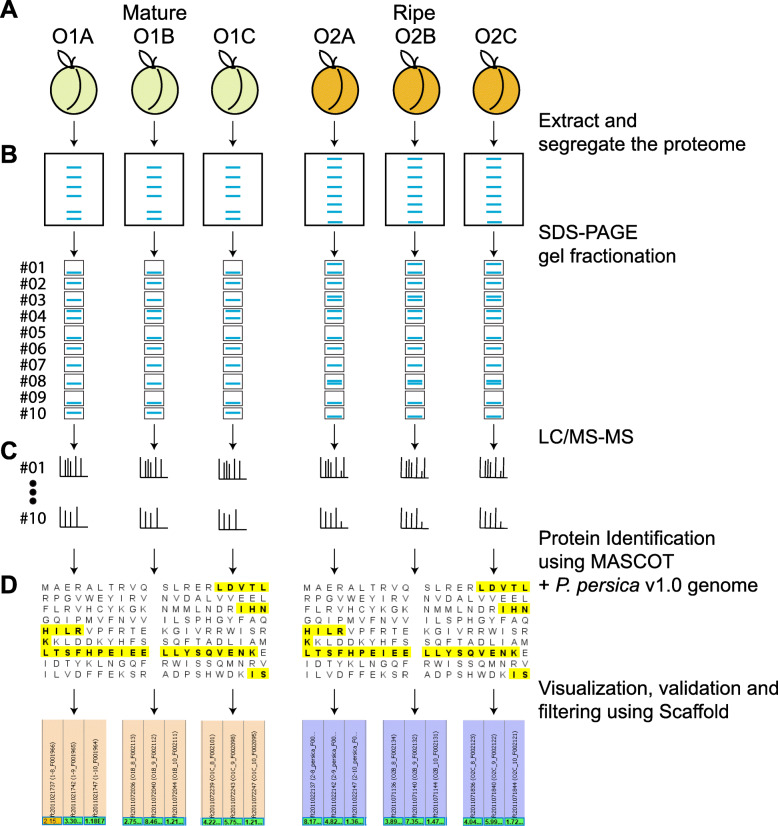
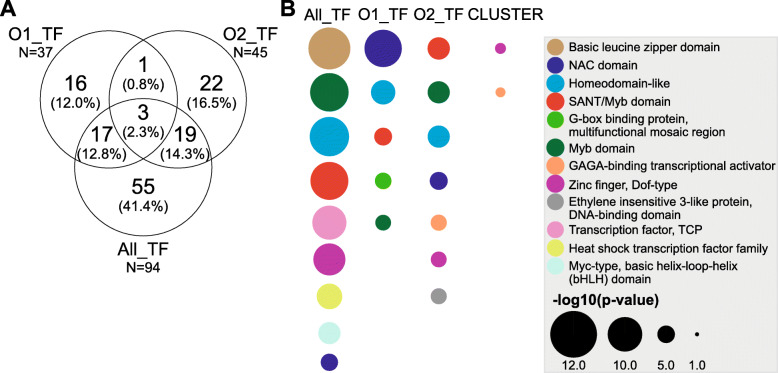
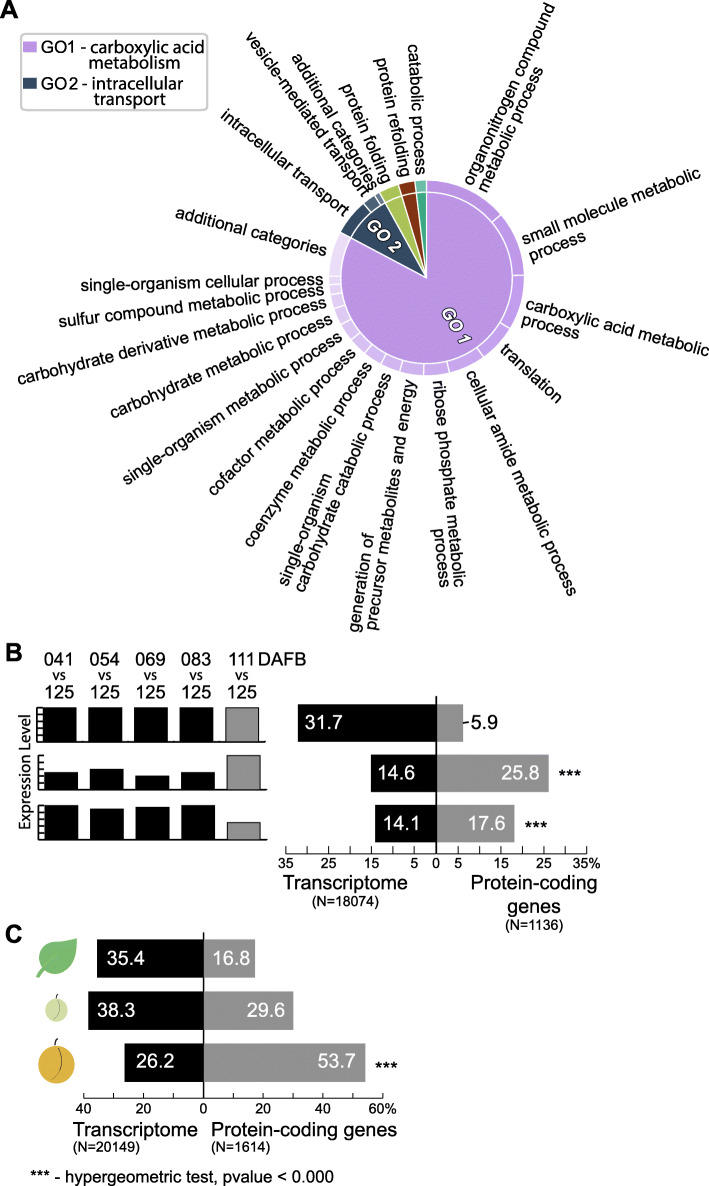
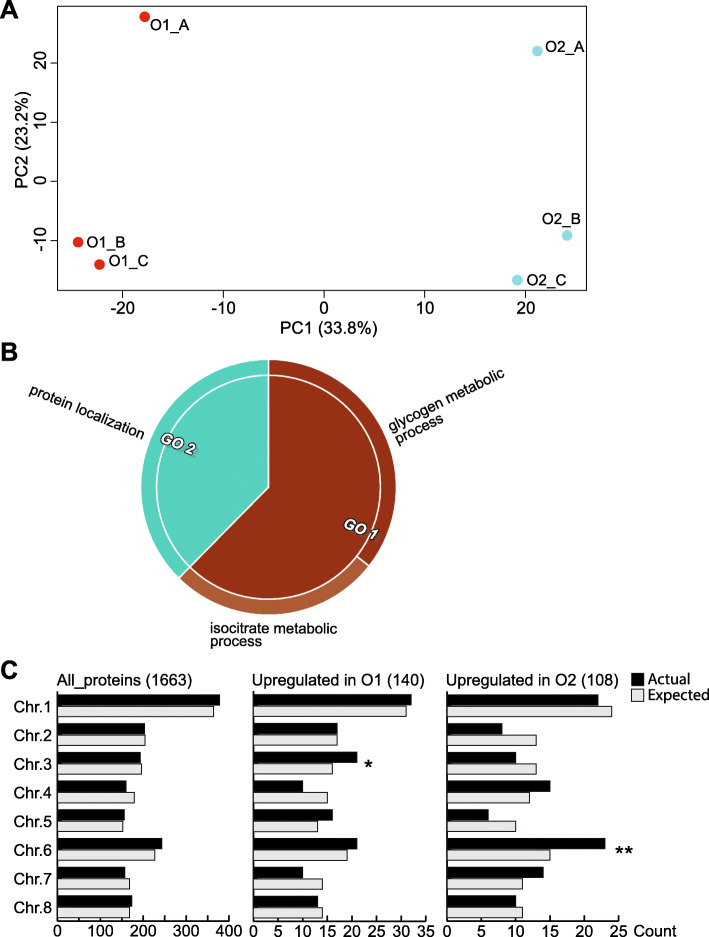
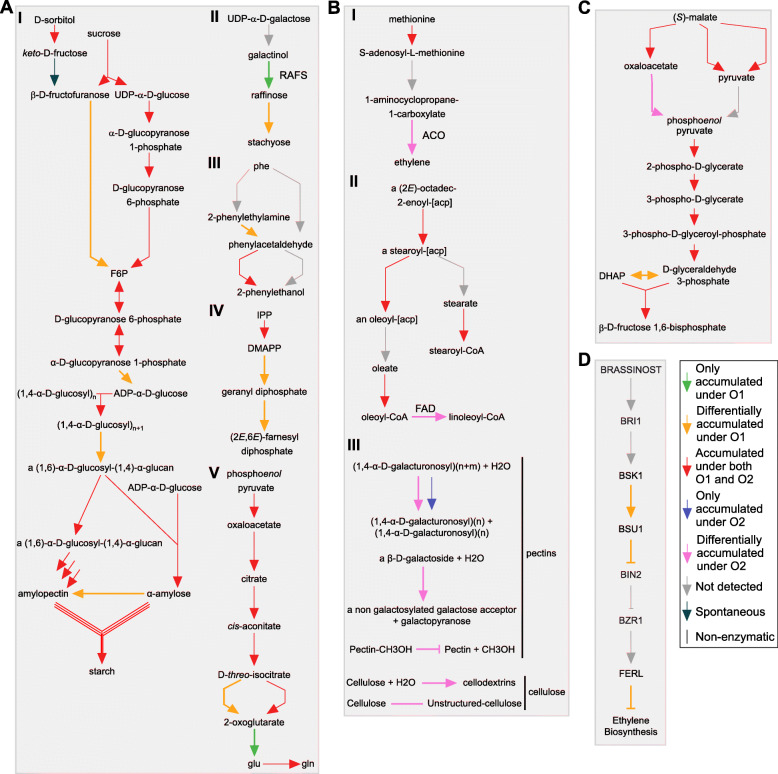
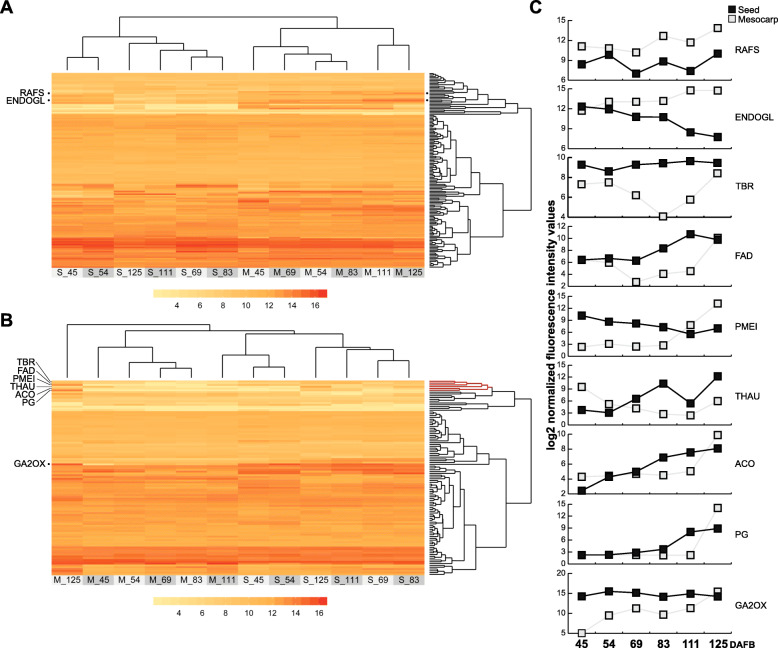
To accomplish this goal, the proteome from mature and ripe fruit was assessed from the variety O'Henry through shotgun proteomics using 1D-gel (PAGE-SDS) as fractionation method followed by LC/MS-MS analysis. Data from the 131,435 spectra could be matched to 2740 proteins, using the peach genome reference v1. After data pre-treatment, 1663 proteins could be used for comparison with datasets assessed using transcriptomic approaches and for quantitative protein accumulation analysis. Close to 26% of the genes that code for the proteins assessed displayed higher expression at ripe fruit compared to other fruit developmental stages, based on published transcriptomic data. Differential accumulation analysis between mature and ripe fruit revealed that 15% of the proteins identified were modulated by the ripening process, with glycogen and isocitrate metabolism, and protein localization overrepresented in mature fruit, as well as cell wall modification in ripe fruit. Potential biomarkers for the ripening process, due to their differential accumulation and gene expression pattern, included a pectin methylesterase inhibitor, a gibbellerin 2-beta-dioxygenase, an omega-6 fatty acid desaturase, a homeobox-leucine zipper protein and an ACC oxidase. Transcription factors enriched in NAC and Myb protein domains would target preferentially the genes encoding proteins more abundant in mature and ripe fruit, respectively.
Shotgun proteomics is an unbiased approach to get deeper into the proteome allowing to detect differences in protein abundance between samples. This technique provided a resolution so that individual gene products could be identified. Many proteins likely involved in cell wall and sugar metabolism, aroma and color, change their abundance during the transition from mature to ripe fruit.
桃溶质型品种果实成熟涉及多种生理变化,这些变化直接影响果实的感官品质和贮藏潜力。通过研究成熟果实和成熟后果肉的蛋白质组差异,有可能突出果实成熟过程中关键的分子过程。
为实现这一目标,采用一维凝胶(PAGE-SDS)作为分离方法,通过鸟枪法蛋白质组学对奥亨利品种成熟果实和成熟后果实的蛋白质组进行了评估,随后进行LC/MS-MS分析。使用桃基因组参考v1,131435个光谱数据可与2740种蛋白质匹配。经过数据预处理后,1663种蛋白质可用于与转录组学方法评估的数据集进行比较,并用于蛋白质积累定量分析。根据已发表的转录组数据,近26%编码所评估蛋白质的基因在成熟后果实中的表达高于其他果实发育阶段。成熟果实和成熟后果实之间的差异积累分析表明,15%的已鉴定蛋白质受成熟过程调控,糖原和异柠檬酸代谢以及蛋白质定位在成熟果实中过度表达,而细胞壁修饰在成熟后果实中过度表达。由于其差异积累和基因表达模式,成熟过程的潜在生物标志物包括果胶甲酯酶抑制剂、赤霉素2-β-双加氧酶、ω-6脂肪酸去饱和酶、同源异型框-亮氨酸拉链蛋白和ACC氧化酶。富含NAC和Myb蛋白结构域的转录因子将分别优先靶向编码在成熟果实和成熟后果实中更丰富蛋白质的基因。
鸟枪法蛋白质组学是一种无偏的方法,可更深入地研究蛋白质组,从而检测样品之间蛋白质丰度的差异。该技术提供了分辨率,以便能够鉴定单个基因产物。许多可能参与细胞壁和糖代谢、香气和颜色的蛋白质在从成熟果实到成熟后果实的转变过程中丰度发生变化。