Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104.
Department of Physiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104.
Proc Natl Acad Sci U S A. 2021 Jan 12;118(2). doi: 10.1073/pnas.2020599118.
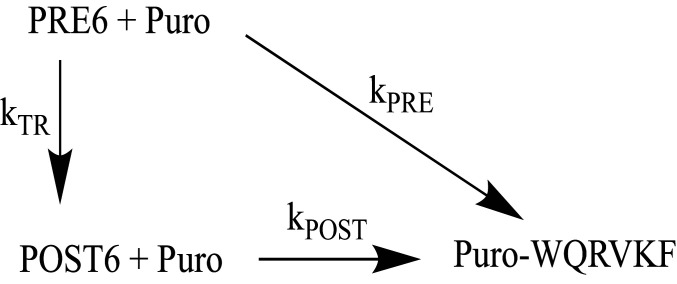
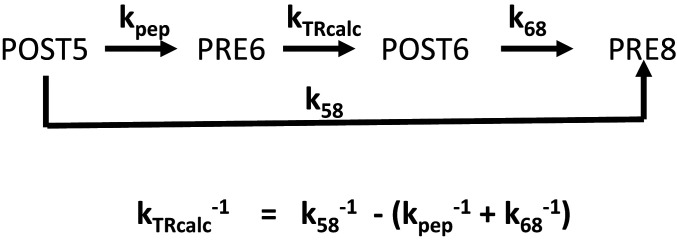
During protein synthesis, nonsense mutations, resulting in premature stop codons (PSCs), produce truncated, inactive protein products. Such defective gene products give rise to many diseases, including cystic fibrosis, Duchenne muscular dystrophy (DMD), and some cancers. Small molecule nonsense suppressors, known as TRIDs (translational read-through-inducing drugs), stimulate stop codon read-through. The best characterized TRIDs are ataluren, which has been approved by the European Medicines Agency for the treatment of DMD, and G418, a structurally dissimilar aminoglycoside. Previously [1], we applied a highly purified in vitro eukaryotic translation system to demonstrate that both aminoglycosides like G418 and more hydrophobic molecules like ataluren stimulate read-through by direct interaction with the cell's protein synthesis machinery. Our results suggested that they might do so by different mechanisms. Here, we pursue this suggestion through a more-detailed investigation of ataluren and G418 effects on read-through. We find that ataluren stimulation of read-through derives exclusively from its ability to inhibit release factor activity. In contrast, G418 increases functional near-cognate tRNA mispairing with a PSC, resulting from binding to its tight site on the ribosome, with little if any effect on release factor activity. The low toxicity of ataluren suggests that development of new TRIDs exclusively directed toward inhibiting termination should be a priority in combatting PSC diseases. Our results also provide rate measurements of some of the elementary steps during the eukaryotic translation elongation cycle, allowing us to determine how these rates are modified when cognate tRNA is replaced by near-cognate tRNA ± TRIDs.
在蛋白质合成过程中,无意义突变导致提前终止密码子(PSCs)产生截短的、无活性的蛋白质产物。这些有缺陷的基因产物会导致许多疾病,包括囊性纤维化、杜氏肌营养不良症(DMD)和一些癌症。小分子无意义抑制物,称为 TRIDs(翻译通读诱导药物),可刺激终止密码子通读。最具代表性的 TRIDs 是 ataluren,它已被欧洲药品管理局批准用于治疗 DMD,以及结构不同的氨基糖苷类抗生素 G418。以前[1],我们应用高度纯化的体外真核翻译系统证明,像 G418 这样的氨基糖苷类抗生素和像 ataluren 这样更疏水的分子通过与细胞的蛋白质合成机制直接相互作用来刺激通读。我们的结果表明,它们可能通过不同的机制起作用。在这里,我们通过更详细地研究 ataluren 和 G418 对通读的影响来探讨这一建议。我们发现,ataluren 对通读的刺激完全来自于它抑制释放因子活性的能力。相比之下,G418 通过结合到核糖体的紧密结合位点,增加了与 PSC 的功能性近同功 tRNA 错配,对释放因子活性的影响很小,如果有的话。ataluren 的低毒性表明,开发专门用于抑制终止的新型 TRIDs 应该是对抗 PSC 疾病的优先事项。我们的结果还提供了真核翻译延伸循环中一些基本步骤的速率测量值,使我们能够确定当同功 tRNA 被近同功 tRNA ± TRIDs 取代时,这些速率如何被修改。