Xia Xiaoting, Zhang Shunjin, Zhang Huaju, Zhang Zijing, Chen Ningbo, Li Zhigang, Sun Hongxia, Liu Xian, Lyu Shijie, Wang Xianwei, Li Zhiming, Yang Peng, Xu Jiawei, Ding Xiaoting, Shi Qiaoting, Wang Eryao, Ru Baorui, Xu Zejun, Lei Chuzhao, Chen Hong, Huang Yongzhen
Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, No. 22 Xinong Road, Yangling, 712100, Shaanxi, China.
Pingdingshan animal husbandry technology promotion station, Pingdingshan, 467000, Henan, China.
BMC Genomics. 2021 Jan 9;22(1):43. doi: 10.1186/s12864-020-07340-0.
Native cattle breeds are an important source of genetic variation because they might carry alleles that enable them to adapt to local environment and tough feeding conditions. Jiaxian Red, a Chinese native cattle breed, is reported to have originated from crossbreeding between taurine and indicine cattle; their history as a draft and meat animal dates back at least 30 years. Using whole-genome sequencing (WGS) data of 30 animals from the core breeding farm, we investigated the genetic diversity, population structure and genomic regions under selection of Jiaxian Red cattle. Furthermore, we used 131 published genomes of world-wide cattle to characterize the genomic variation of Jiaxian Red cattle.
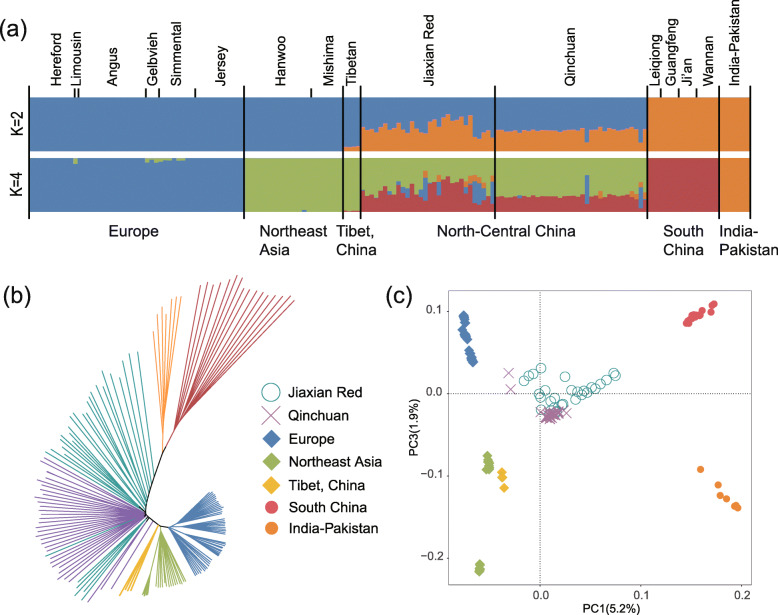
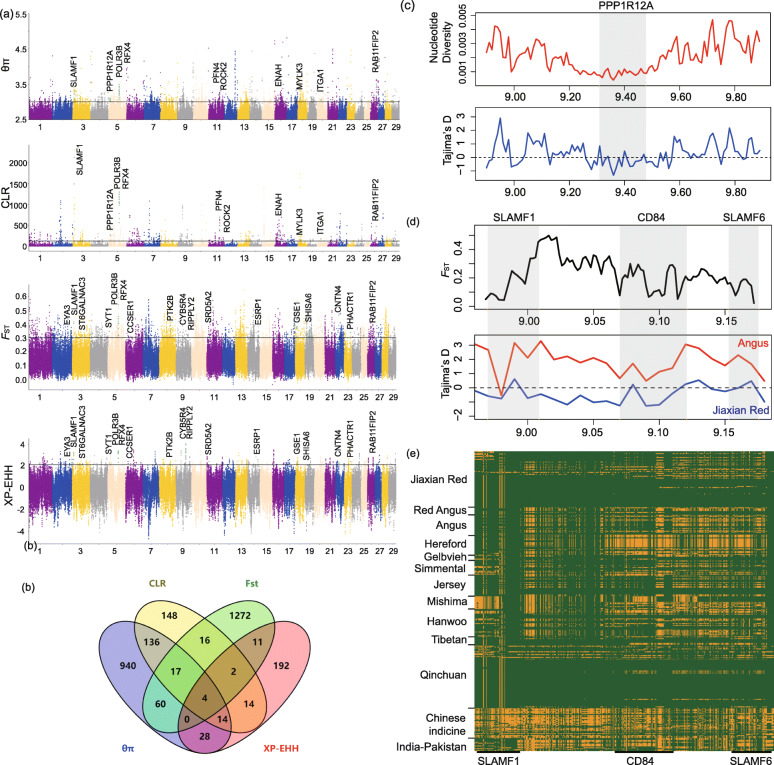
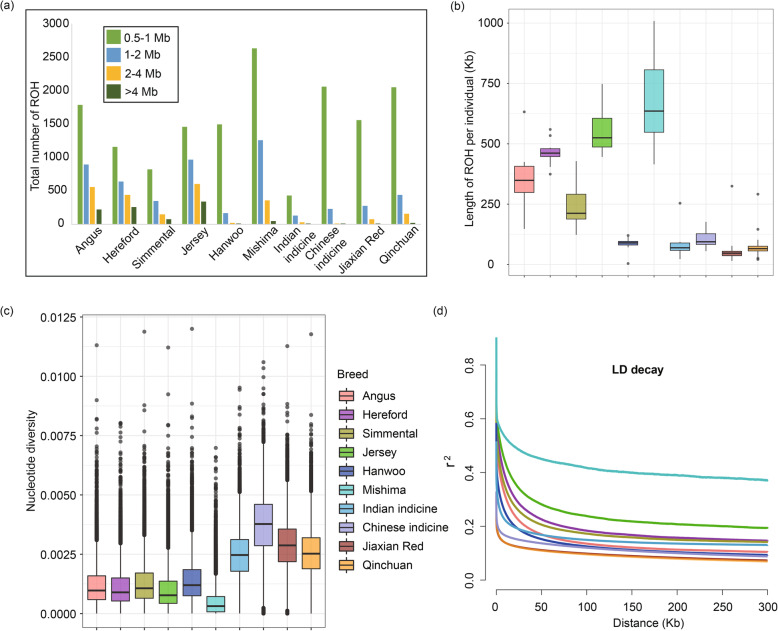
The population structure analysis revealed that Jiaxian Red cattle harboured the ancestry with East Asian taurine (0.493), Chinese indicine (0.379), European taurine (0.095) and Indian indicine (0.033). Three methods (nucleotide diversity, linkage disequilibrium decay and runs of homozygosity) implied the relatively high genomic diversity in Jiaxian Red cattle. We used θπ, CLR, F and XP-EHH methods to look for the candidate signatures of positive selection in Jiaxian Red cattle. A total number of 171 (θπ and CLR) and 17 (F and XP-EHH) shared genes were identified using different detection strategies. Functional annotation analysis revealed that these genes are potentially responsible for growth and feed efficiency (CCSER1), meat quality traits (ROCK2, PPP1R12A, CYB5R4, EYA3, PHACTR1), fertility (RFX4, SRD5A2) and immune system response (SLAMF1, CD84 and SLAMF6).
We provide a comprehensive overview of sequence variations in Jiaxian Red cattle genomes. Selection signatures were detected in genomic regions that are possibly related to economically important traits in Jiaxian Red cattle. We observed a high level of genomic diversity and low inbreeding in Jiaxian Red cattle. These results provide a basis for further resource protection and breeding improvement of this breed.
本地牛品种是遗传变异的重要来源,因为它们可能携带使其能够适应当地环境和恶劣饲养条件的等位基因。据报道,中国本地牛品种郏县红牛起源于普通牛和瘤牛的杂交;其作为役用和肉用动物的历史至少可追溯到30年前。利用核心育种场30头牛的全基因组测序(WGS)数据,我们研究了郏县红牛的遗传多样性、群体结构和选择下的基因组区域。此外,我们使用131个已发表的全球牛基因组来表征郏县红牛的基因组变异。
群体结构分析表明,郏县红牛含有东亚普通牛(0.493)、中国瘤牛(0.379)、欧洲普通牛(0.095)和印度瘤牛(0.033)的血统。三种方法(核苷酸多样性、连锁不平衡衰减和纯合子片段)表明郏县红牛具有相对较高的基因组多样性。我们使用θπ、CLR、F和XP-EHH方法寻找郏县红牛正选择的候选特征。使用不同的检测策略共鉴定出171个(θπ和CLR)和17个(F和XP-EHH)共享基因。功能注释分析表明,这些基因可能与生长和饲料效率(CCSER1)、肉质性状(ROCK2、PPP1R12A、CYB5R4、EYA3、PHACTR1)、繁殖力(RFX4、SRD5A2)和免疫系统反应(SLAMF1、CD84和SLAMF6)有关。
我们提供了郏县红牛基因组序列变异的全面概述。在可能与郏县红牛重要经济性状相关的基因组区域检测到了选择特征。我们观察到郏县红牛具有高水平的基因组多样性和低近亲繁殖率。这些结果为该品种的进一步资源保护和育种改良提供了依据。