Sagi Satyanarayana V, Joshi Hareesh, Trotman Jamie, Elsey Terence, Swamy Ashwini, Rajkanna Jeyanthy, Bhat Nazir A, Haddadin Firas J S, Oyibo Samson O, Park Soo-Mi
Department of Diabetes and Endocrinology, Peterborough City Hospital, Peterborough, UK.
East Midlands and East of England NHS Genomic Laboratory Hub, Cambridge University Hospital NHS Foundation Trust, Cambridge Biomedical Campus, Cambridge, UK.
Endocrinol Diabetes Metab Case Rep. 2020 Sep 23;2020. doi: 10.1530/EDM-20-0084.
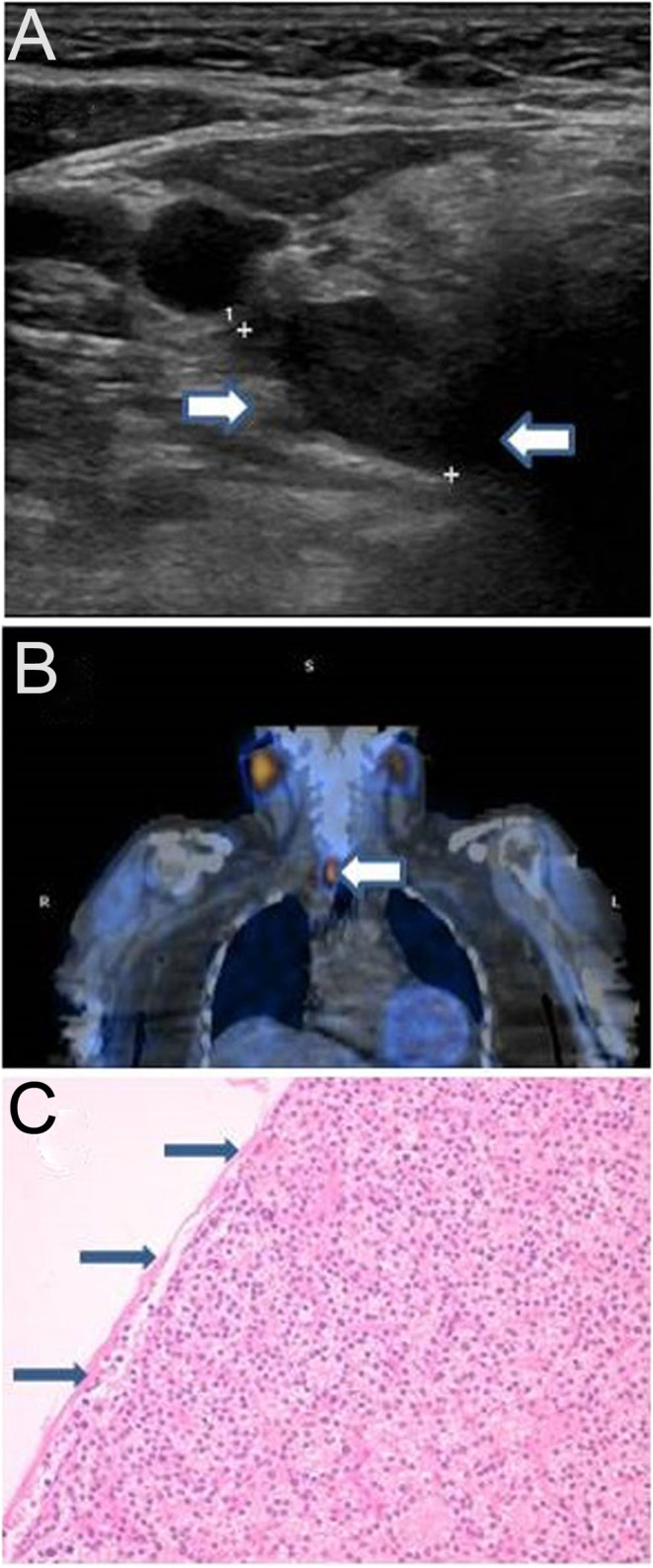
Familial hypocalciuric hypercalcaemia (FHH) is a dominantly inherited, lifelong benign disorder characterised by asymptomatic hypercalcaemia, relative hypocalciuria and variable parathyroid hormone levels. It is caused by loss-of-function pathogenic variants in the calcium-sensing receptor (CASR) gene. Primary hyperparathyroidism (PHPT) is characterised by variable hypercalcaemia in the context of non-suppressed parathyroid hormone levels. Unlike patients with FHH, patients with severe hypercalcaemia due to PHPT are usually symptomatic and are at risk of end-organ damage affecting the kidneys, bone, heart, gastrointestinal system and CNS. Surgical resection of the offending parathyroid gland(s) is the treatment of choice for PHPT, while dietary adjustment and reassurance is the mainstay of management for patients with FHH. The occurrence of both FHH and primary hyperparathyroidism (PHPT) in the same patient has been described. We report an interesting case of FHH due to a novel CASR variant confirmed in a mother and her two daughters and the possible coexistence of FHH and PHPT in the mother, highlighting the challenges involved in diagnosis and management.
Familial hypocalciuric hypercalcaemia (FHH) and primary hyperparathyroidism (PHPT) can coexist in the same patient. Urinary calcium creatinine clearance ratio can play a role in distinguishing between PHPT and FHH. Genetic testing should be considered in managing patients with PHPT and FHH where the benefit may extend to the wider family. Family segregation studies can play an important role in the reclassification of variants of uncertain significance. Parathyroidectomy has no benefit in patients with FHH and therefore, it is important to exclude FHH prior to considering surgery. For patients with coexisting FHH and PHPT, parathyroidectomy will reduce the risk of complications from the severe hypercalcaemia associated with PHPT.
家族性低钙血症性高钙血症(FHH)是一种常染色体显性遗传的终身性良性疾病,其特征为无症状性高钙血症、相对低钙尿症以及甲状旁腺激素水平多变。它由钙敏感受体(CASR)基因的功能丧失性致病变异引起。原发性甲状旁腺功能亢进症(PHPT)的特征是在甲状旁腺激素水平未被抑制的情况下出现多变的高钙血症。与FHH患者不同,因PHPT导致严重高钙血症的患者通常有症状,且有发生影响肾脏、骨骼、心脏、胃肠道系统和中枢神经系统的终末器官损害的风险。手术切除有问题的甲状旁腺是PHPT的首选治疗方法,而饮食调整和安慰是FHH患者管理的主要手段。同一患者同时发生FHH和原发性甲状旁腺功能亢进症(PHPT)的情况已有报道。我们报告了一例有趣的FHH病例,在一位母亲及其两个女儿中发现了一种新的CASR变异,且母亲可能同时存在FHH和PHPT,突出了诊断和管理中所涉及的挑战。
家族性低钙血症性高钙血症(FHH)和原发性甲状旁腺功能亢进症(PHPT)可在同一患者中共存。尿钙肌酐清除率在区分PHPT和FHH方面可发挥作用。在管理PHPT和FHH患者时应考虑进行基因检测,其益处可能会惠及更广泛的家族。家系分离研究在对意义未明的变异进行重新分类方面可发挥重要作用。甲状旁腺切除术对FHH患者无益,因此,在考虑手术前排除FHH很重要。对于同时存在FHH和PHPT的患者,甲状旁腺切除术将降低与PHPT相关的严重高钙血症并发症的风险。