Yang Rong, Zhou Yuwei, Du Chengli, Wu Yihe
Department of Radiology, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Department of Thoracic Surgery, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
J Thorac Dis. 2020 Dec;12(12):7355-7364. doi: 10.21037/jtd-20-3453.
Lung adenocarcinoma is the main pathological type of non-small cell lung cancer (NSCLC). In this study, we analyzed the gene expression profile of lung adenocarcinoma tumor and paracancerous tissues by bioinformatics to assess the genes and signal pathways related to lung adenocarcinoma.
The expression data of GSE7670, GSE27262, and GSE32863 were downloaded from the Gene Expression Omnibus (GEO) database. The three microarray data sets were integrated to obtain common differential expression genes of lung adenocarcinoma tumor and adjacent tissues. The STRING database was used to construct the protein-protein interaction (PPI) network of lung adenocarcinoma and mine the gene modules and core genes in the network, and the online tools, GEPIA and Kaplan-Meier plotter were used to further verify and analyze the core genes.
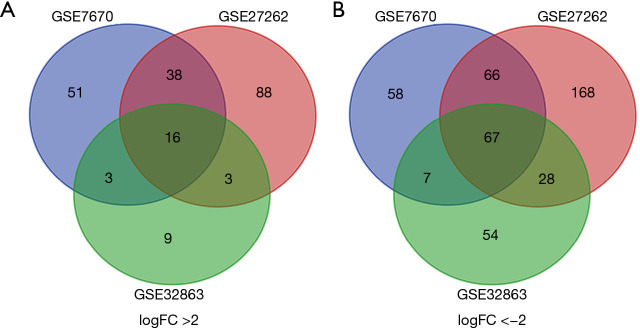
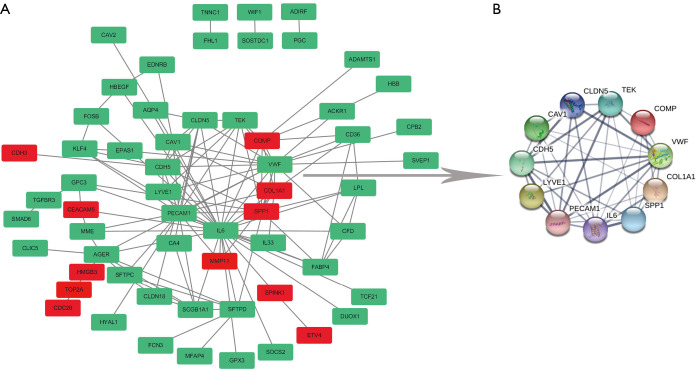
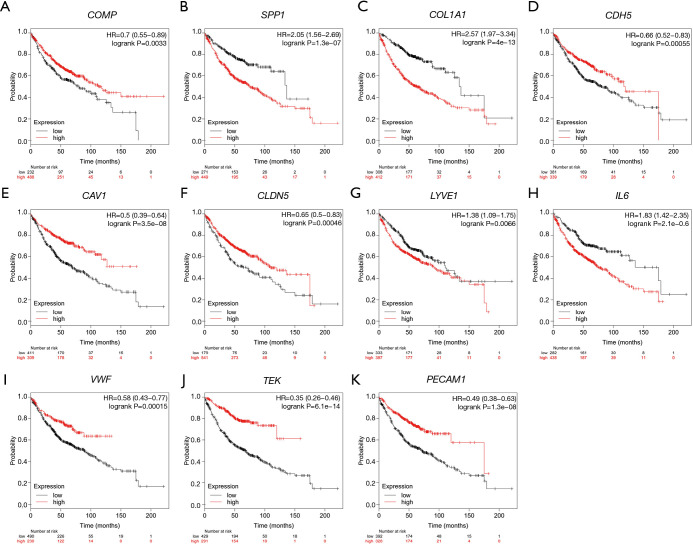
There were 109 pairs of lung adenocarcinoma tissues and matched paracancerous normal lung tissues in the three data sets. Eighty-three differentially expressed genes were identified, including 16 up-regulated and 67 down-regulated genes, and 60 differentially expressed genes were successfully incorporated into the PPI network complex. Eleven core genes were identified in the PPI network complex, including three up-regulated (, , ) and eight down-regulated genes (, , , , , , , ). These core genes were verified by the GEPIA tumor database. Survival analysis showed that expression of the core genes was significantly related to the prognosis of lung adenocarcinoma. KEGG pathway analysis of core genes showed six genes (, , , , , ) were significantly enriched in the PI3K-Akt signaling-pathway (P=1.62E-06).
By analyzing the differential expression genes of lung adenocarcinoma and paracancerous normal tissues with bioinformatics, 11 genes with significant differential expression and significant influence on prognosis were identified. The findings may provide new concepts for developing diagnosis and treatment targets and prognosis markers for lung adenocarcinoma.
肺腺癌是非小细胞肺癌(NSCLC)的主要病理类型。在本研究中,我们通过生物信息学分析肺腺癌肿瘤组织和癌旁组织的基因表达谱,以评估与肺腺癌相关的基因和信号通路。
从基因表达综合数据库(GEO)下载GSE7670、GSE27262和GSE32863的表达数据。整合这三个微阵列数据集以获得肺腺癌肿瘤组织和相邻组织的共同差异表达基因。使用STRING数据库构建肺腺癌的蛋白质-蛋白质相互作用(PPI)网络,并挖掘网络中的基因模块和核心基因,使用在线工具GEPIA和Kaplan-Meier绘图仪对核心基因进行进一步验证和分析。
三个数据集中共有109对肺腺癌组织及匹配的癌旁正常肺组织。鉴定出83个差异表达基因,其中16个上调基因和67个下调基因,60个差异表达基因成功纳入PPI网络复合体。在PPI网络复合体中鉴定出11个核心基因,包括3个上调基因(、、)和8个下调基因(、、、、、、、)。这些核心基因通过GEPIA肿瘤数据库得到验证。生存分析表明,核心基因的表达与肺腺癌的预后显著相关。对核心基因的KEGG通路分析表明,6个基因(、、、、、)在PI3K-Akt信号通路中显著富集(P = 1.62E-06)。
通过生物信息学分析肺腺癌与癌旁正常组织的差异表达基因,鉴定出11个差异表达显著且对预后有显著影响的基因。这些发现可能为开发肺腺癌的诊断和治疗靶点及预后标志物提供新的思路。