Department of Respiratory Medicine, The First Hospital of Changsha, Changsha, China.
Department of Cardiology, The First Hospital of Changsha, Changsha, China.
PeerJ. 2022 Jan 31;10:e12731. doi: 10.7717/peerj.12731. eCollection 2022.
Identification of accurate prognostic biomarkers is still particularly urgent for improving the poor survival of lung cancer patients. In this study, we aimed to identity the potential biomarkers in Chinese lung cancer population via bioinformatics analysis.
In this study, the differentially expressed genes (DEGs) in lung cancer were identified using six datasets from Gene Expression Omnibus (GEO) database. Subsequently, enrichment analysis was conducted to evaluate the underlying molecular mechanisms involved in progression of lung cancer. Protein-protein interaction (PPI) and CytoHubba analysis were performed to determine the hub genes. The GEPIA, Human Protein Atlas (HPA), Kaplan-Meier plotter, and TIMER databases were used to explore the hub genes. The receiver operating characteristic (ROC) analysis was performed to evaluate the diagnostic value of hub genes. Reverse transcription quantitative PCR (qRT-PCR) was used to validate the expression levels of hub genes in 10 pairs of lung cancer paired tissues.
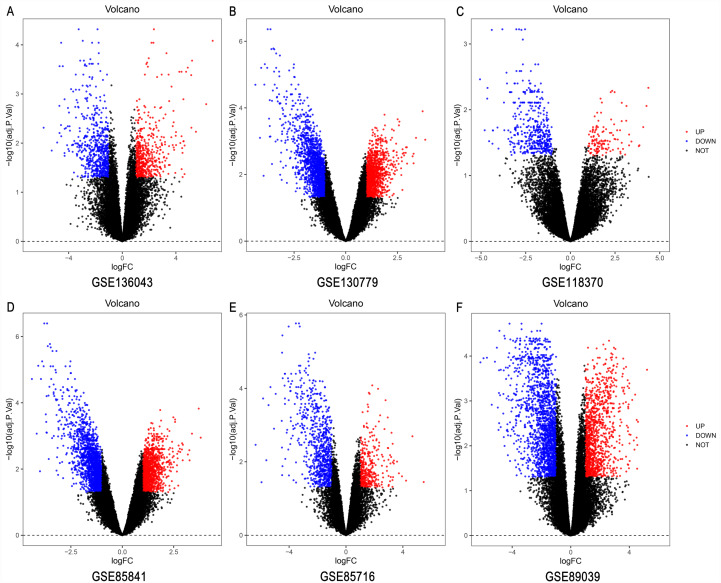
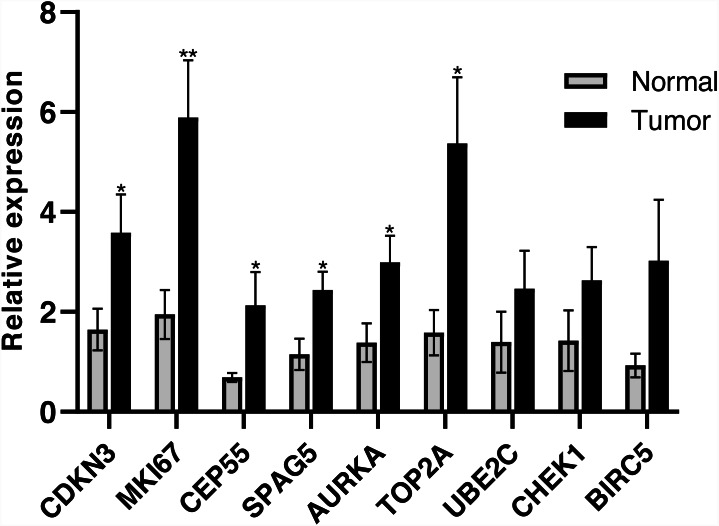
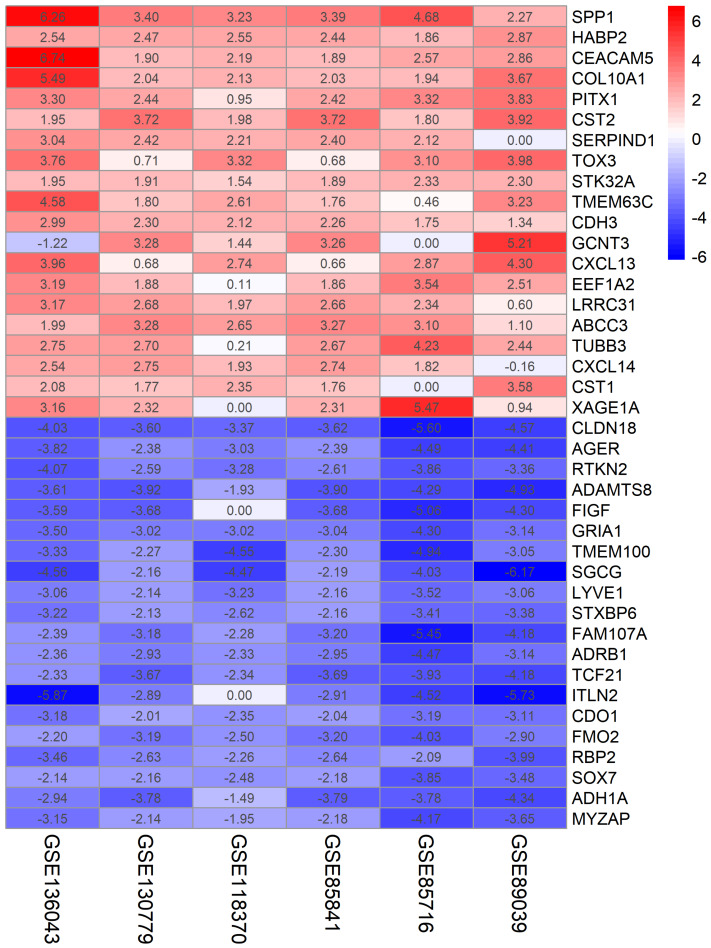
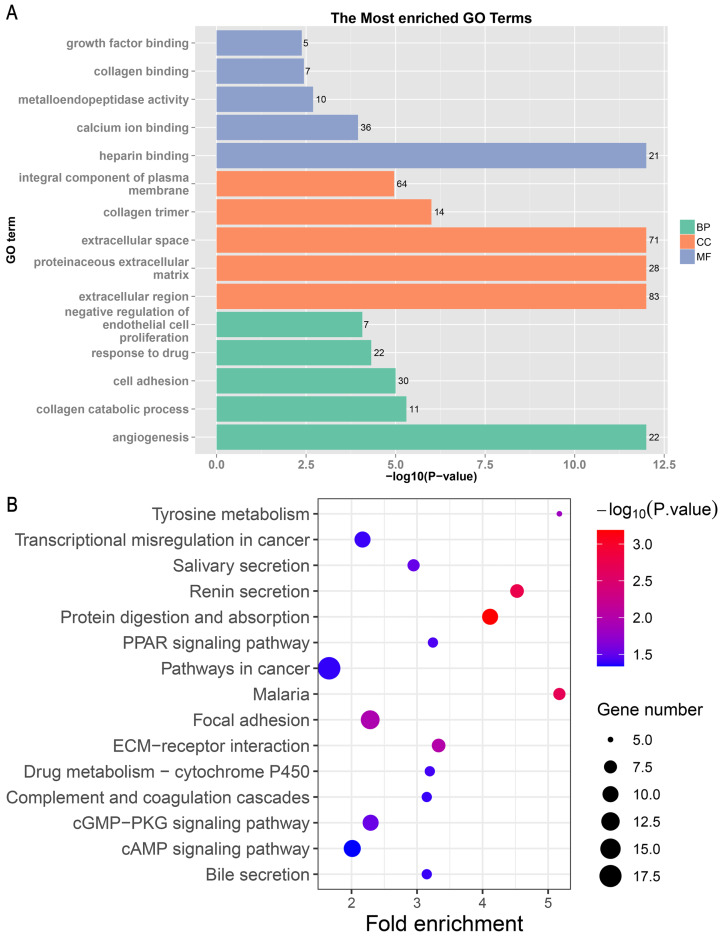
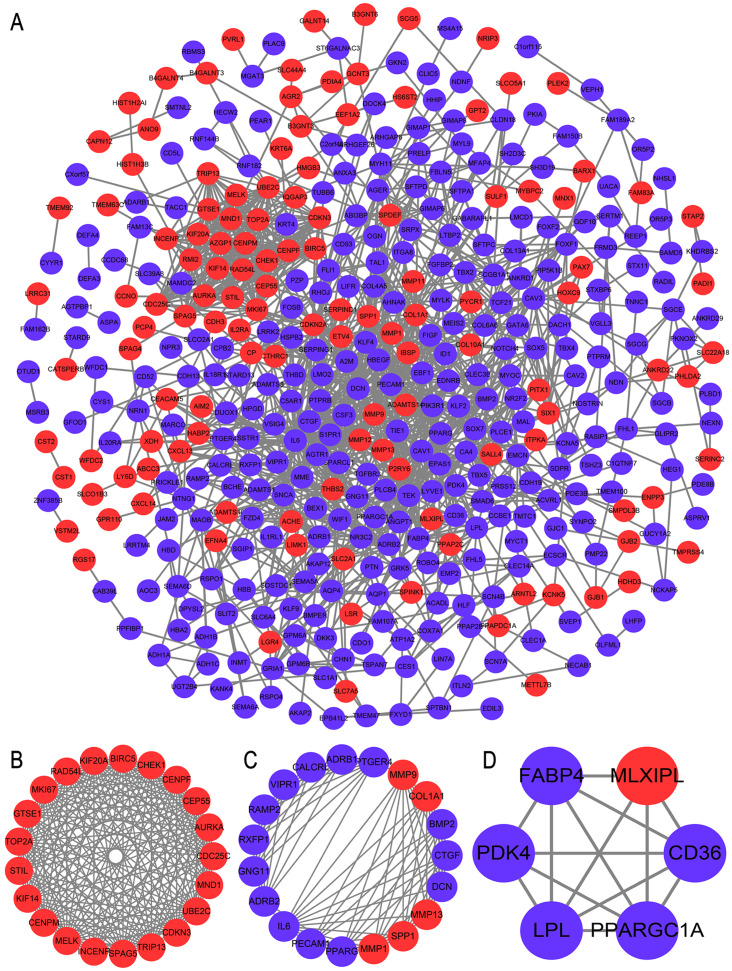
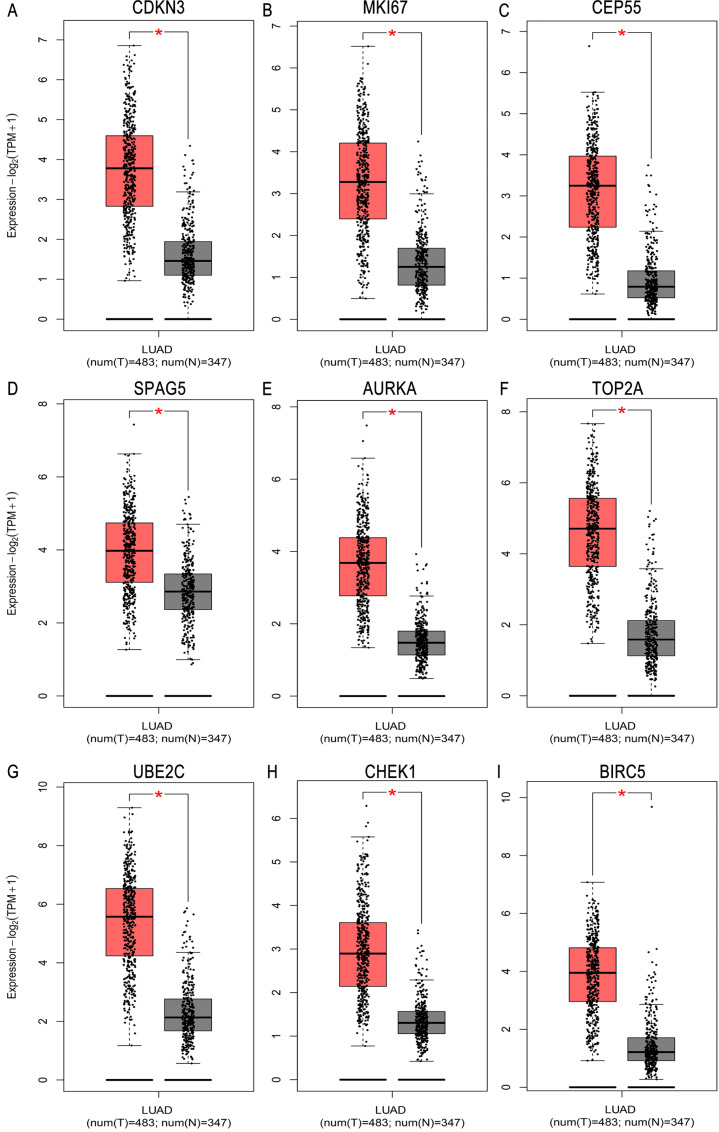
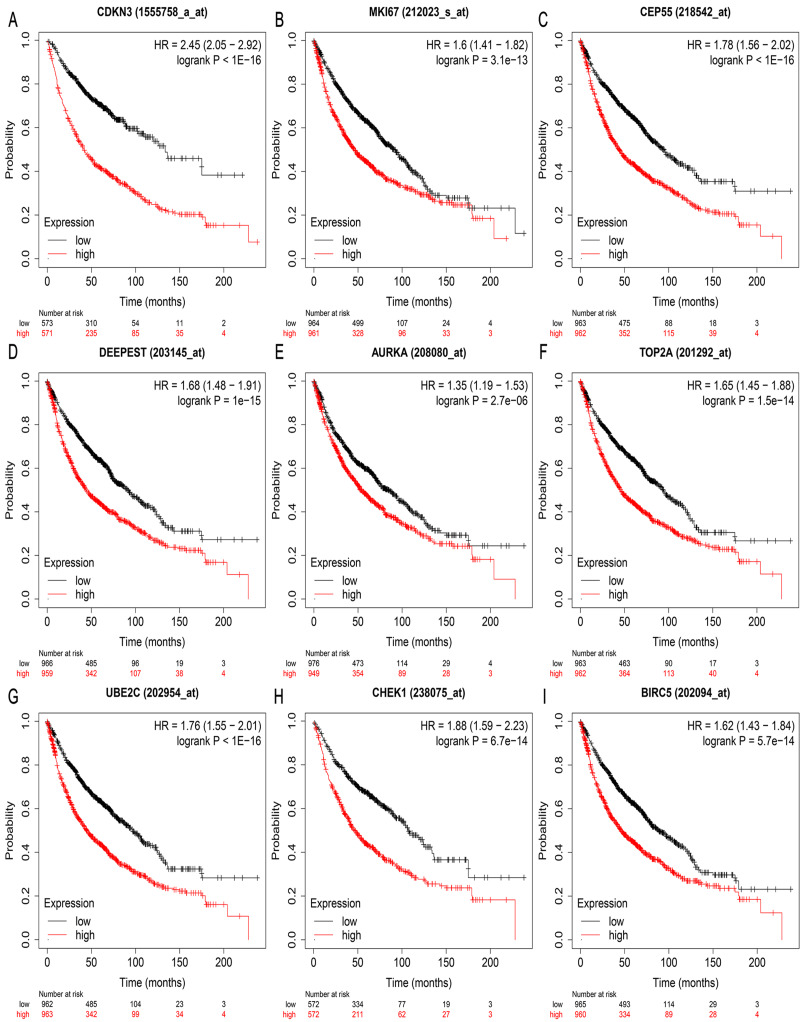
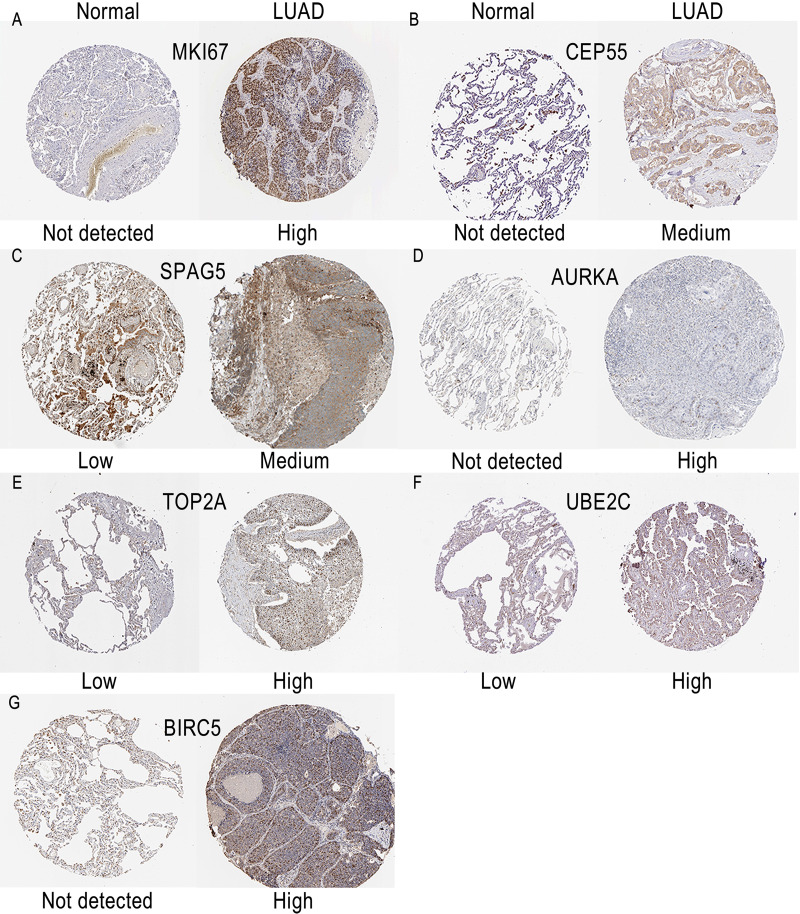
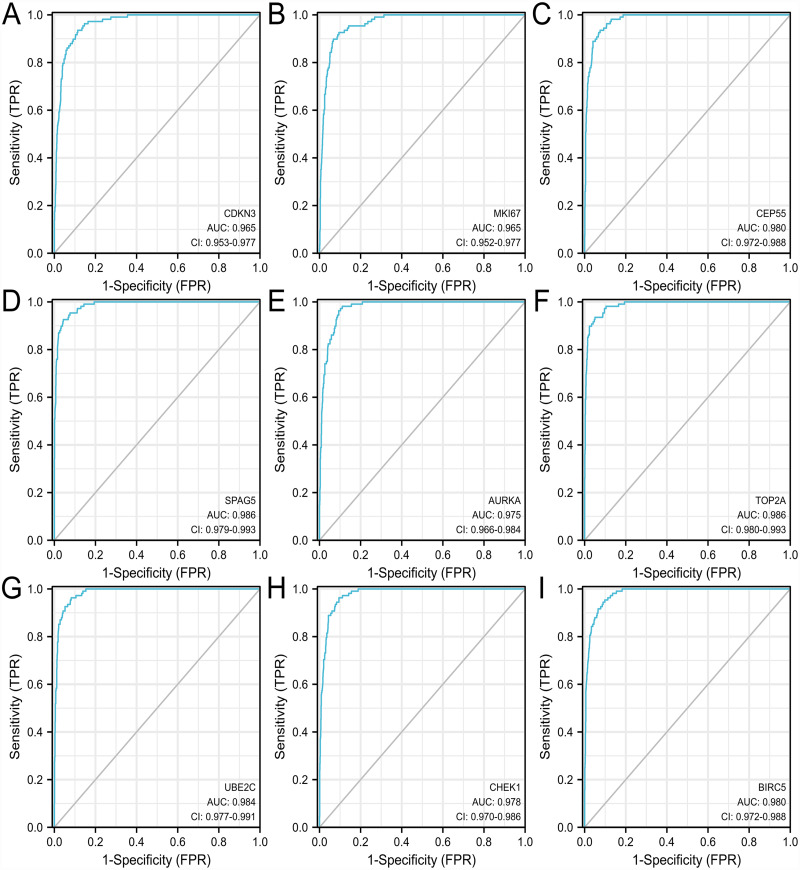
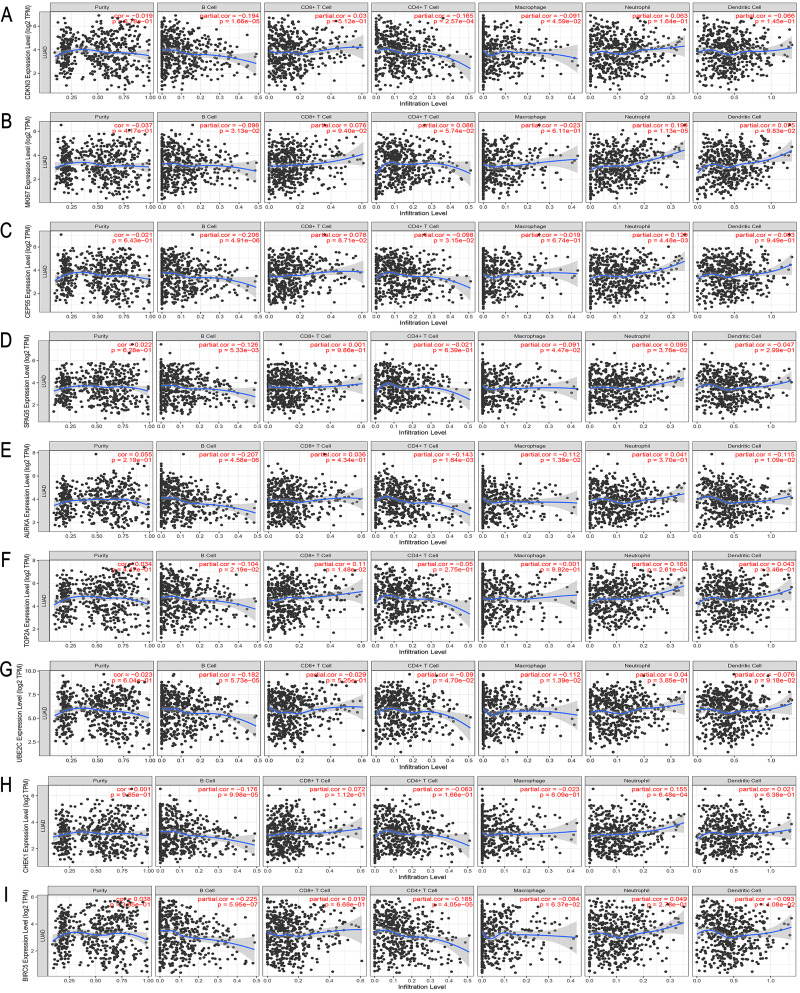
A total of 499 overlapping DEGs (160 upregulated and 339 downregulated genes) were identified in the microarray datasets. DEGs were mainly associated with pathways in cancer, focal adhesion, and protein digestion and absorption. There were nine hub genes (CDKN3, MKI67, CEP55, SPAG5, AURKA, TOP2A, UBE2C, CHEK1 and BIRC5) identified by PPI and module analysis. In GEPIA database, the expression levels of these genes in lung cancer tissues were significantly upregulated compared with normal lung tissues. The results of prognostic analysis showed that relatively higher expression of hub genes was associated with poor prognosis of lung cancer. In HPA database, most hub genes were highly expressed in lung cancer tissues. The hub genes have good diagnostic efficiency in lung cancer and normal tissues. The expression of any hub gene was associated with the infiltration of at least two immune cells. qRT-PCR confirmed that the expression level of CDKN3, MKI67, CEP55, SPAG5, AURKA, TOP2A were highly expressed in lung cancer tissues.
The hub genes and functional pathways identified in this study may contribute to understand the molecular mechanisms of lung cancer. Our findings may provide new therapeutic targets for lung cancer patients.
对于提高肺癌患者的生存率,识别准确的预后生物标志物仍然尤为迫切。在这项研究中,我们旨在通过生物信息学分析鉴定中国肺癌人群中的潜在生物标志物。
本研究通过从基因表达综合数据库(GEO)数据库中获取的六个数据集,鉴定肺癌中的差异表达基因(DEGs)。随后,进行富集分析以评估涉及肺癌进展的潜在分子机制。进行蛋白质-蛋白质相互作用(PPI)和 CytoHubba 分析以确定关键基因。使用 GEPIA、人类蛋白质图谱(HPA)、Kaplan-Meier plotter 和 TIMER 数据库来探索关键基因。进行受试者工作特征(ROC)分析以评估关键基因的诊断价值。使用反转录定量 PCR(qRT-PCR)验证 10 对肺癌配对组织中关键基因的表达水平。
在微阵列数据集中共鉴定出 499 个重叠的 DEGs(160 个上调和 339 个下调基因)。DEGs 主要与癌症途径、黏附斑和蛋白消化吸收相关。通过 PPI 和模块分析,鉴定出 9 个关键基因(CDKN3、MKI67、CEP55、SPAG5、AURKA、TOP2A、UBE2C、CHEK1 和 BIRC5)。在 GEPIA 数据库中,这些基因在肺癌组织中的表达水平明显高于正常肺组织。预后分析结果表明,关键基因的相对高表达与肺癌的不良预后相关。在 HPA 数据库中,大多数关键基因在肺癌组织中高表达。关键基因在肺癌和正常组织中具有良好的诊断效率。任何关键基因的表达均与至少两种免疫细胞的浸润有关。qRT-PCR 证实 CDKN3、MKI67、CEP55、SPAG5、AURKA、TOP2A 在肺癌组织中的表达水平较高。
本研究中鉴定的关键基因和功能途径可能有助于理解肺癌的分子机制。我们的研究结果可为肺癌患者提供新的治疗靶点。