Tanawattanasuntorn Tanotnon, Thongpanchang Tienthong, Rungrotmongkol Thanyada, Hanpaibool Chonnikan, Graidist Potchanapond, Tipmanee Varomyalin
Department of Biomedical Sciences and Biomedical Engineering, Faculty of Medicine, Prince of Songkla University, Hat Yai, Songkhla 90110, Thailand.
Department of Chemistry, Faculty of Science and Center of Excellence for Innovation in Chemistry, Mahidol University, Bangkok 10400, Thailand.
ACS Omega. 2020 Dec 21;6(1):606-614. doi: 10.1021/acsomega.0c05102. eCollection 2021 Jan 12.
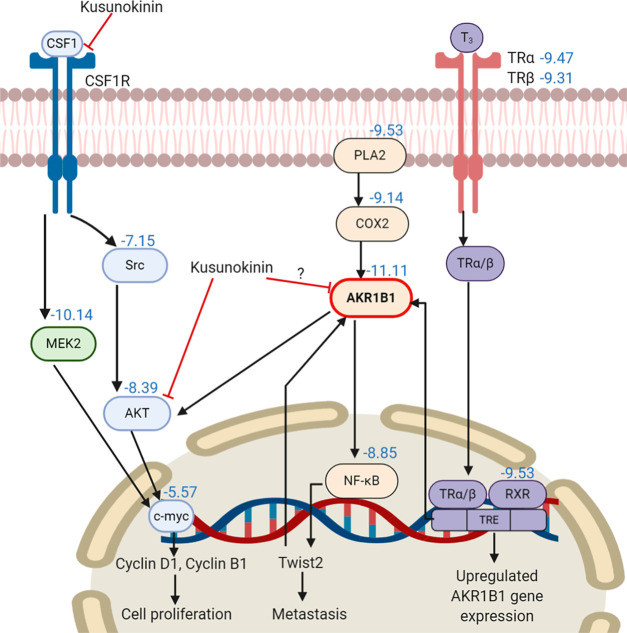
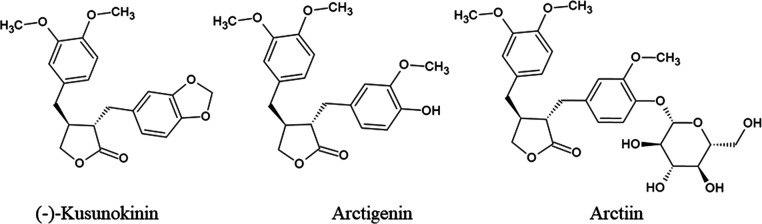
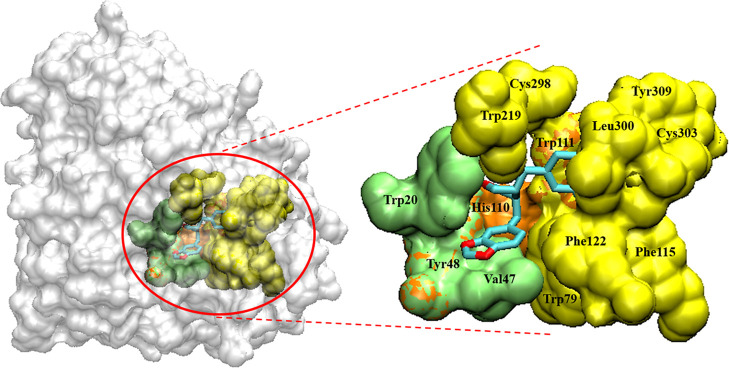
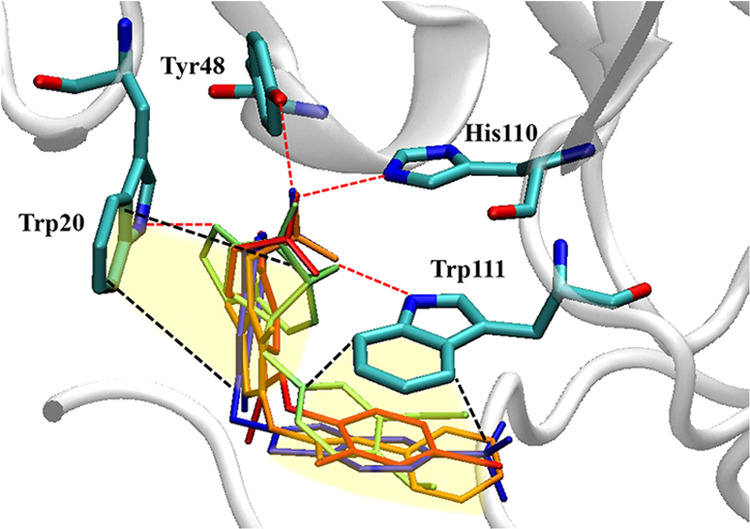
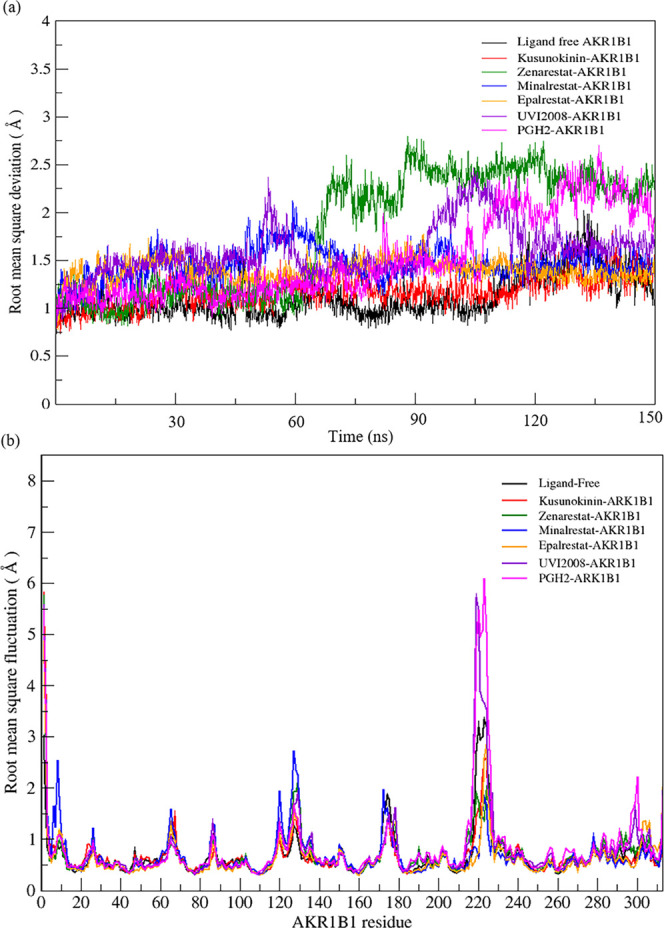
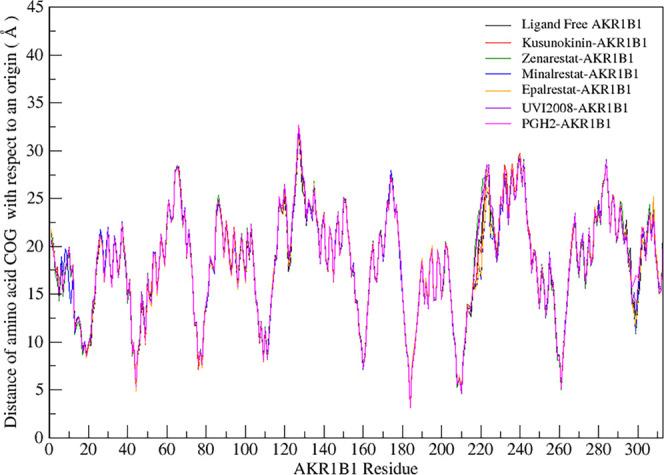
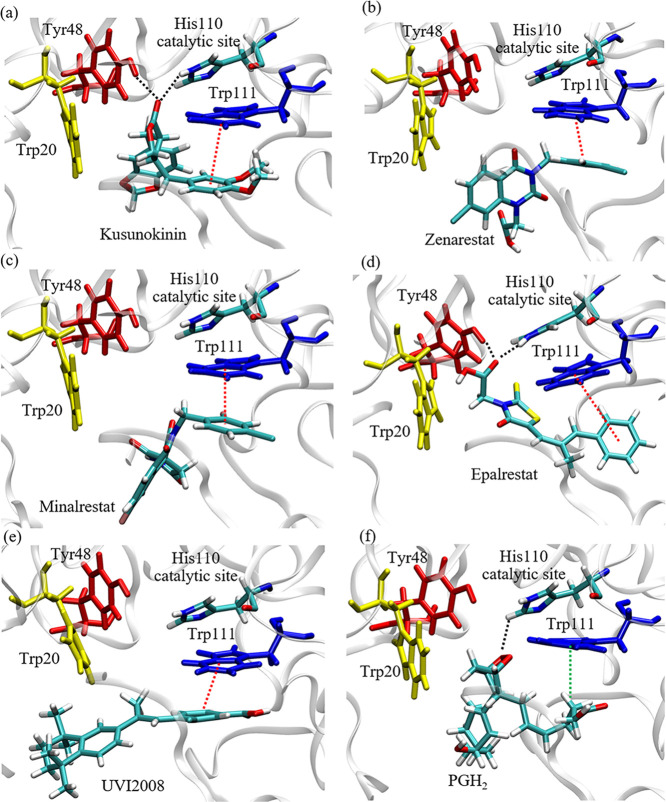
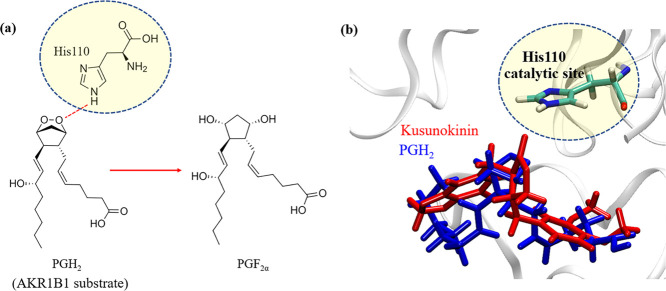
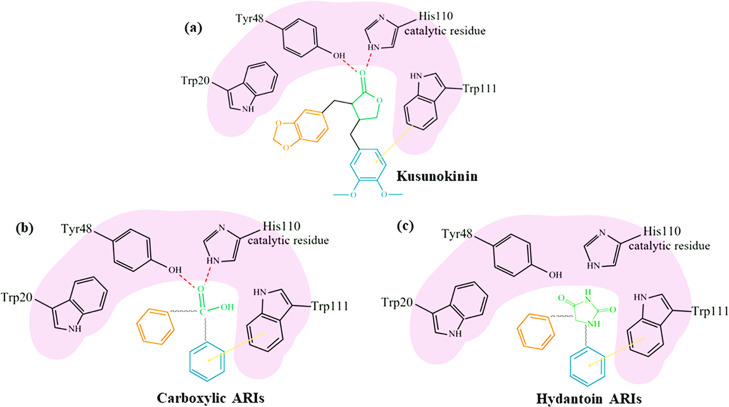
(-)-Kusunokinin performed its anticancer potency through CFS1R and AKT pathways. Its ambiguous binding target has, however, hindered the next development phase. Our study thus applied molecular docking and molecular dynamics simulation to predict the protein target from the pathways. Among various candidates, aldo-keto reductase family 1 member B1 (AKR1B1) was finally identified as a (-)-kusunokinin receptor. The predicted binding affinity of (-)-kusunokinin was better than the selected aldose reductase inhibitors (ARIs) and substrates. The compound also had no significant effect on AKR1B1 conformation. An intriguing AKR1B1 efficacy, with respect to the known inhibitors (epalrestat, zenarestat, and minalrestat) and substrates (UVI2008 and prostaglandin H), as well as a similar interactive insight of the enzyme pocket, pinpointed an ARI equivalence of (-)-kusunokinin. An aromatic ring and a γ-butyrolactone ring shared a role with structural counterparts in known inhibitors. The modeling explained that the aromatic constituent contributed to π-π attraction with Trp111. In addition, the γ-butyrolactone ring bound the catalytic His110 using hydrogen bonds, which could lead to enzymatic inhibition as a consequence of substrate competitiveness. Our computer-based findings suggested that the potential of (-)-kusunokinin could be furthered by in vitro and/or in vivo experiments to consolidate (-)-kusunokinin as a new AKR1B1 antagonist in the future.
(-)-草野宁通过CFS1R和AKT途径发挥其抗癌效力。然而,其模糊的结合靶点阻碍了下一阶段的研发。因此,我们的研究应用分子对接和分子动力学模拟从这些途径中预测蛋白质靶点。在各种候选物中,醛糖酮还原酶家族1成员B1(AKR1B1)最终被确定为(-)-草野宁的受体。(-)-草野宁预测的结合亲和力优于所选的醛糖还原酶抑制剂(ARIs)和底物。该化合物对AKR1B1的构象也没有显著影响。关于已知抑制剂(依帕司他、泽那司他和米那司他)和底物(UVI2008和前列腺素H),AKR1B1具有有趣的功效,以及酶口袋类似的相互作用见解,明确了(-)-草野宁与ARIs相当。一个芳香环和一个γ-丁内酯环与已知抑制剂中的结构对应部分发挥相同作用。该模型解释说,芳香成分有助于与Trp111形成π-π吸引力。此外,γ-丁内酯环通过氢键与催化性的His110结合,这可能由于底物竞争而导致酶抑制。我们基于计算机的研究结果表明,(-)-草野宁的潜力可以通过体外和/或体内实验进一步挖掘,以便未来将(-)-草野宁巩固为一种新的AKR1B1拮抗剂。