Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
Department of Translational Medical Sciences, Section of Pediatrics, Federico II University, Naples, Italy.
EMBO Mol Med. 2021 Feb 5;13(2):e12836. doi: 10.15252/emmm.202012836. Epub 2021 Jan 18.
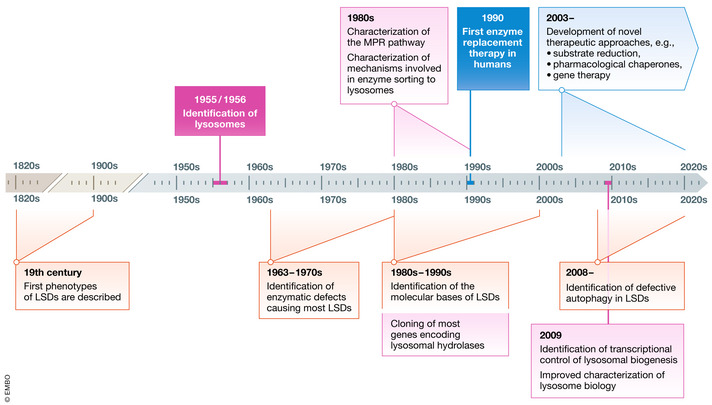
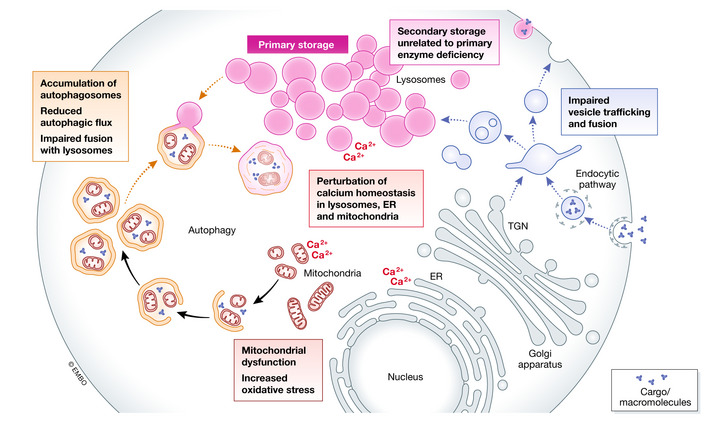
Lysosomal storage diseases are a group of metabolic disorders caused by deficiencies of several components of lysosomal function. Most commonly affected are lysosomal hydrolases, which are involved in the breakdown and recycling of a variety of complex molecules and cellular structures. The understanding of lysosomal biology has progressively improved over time. Lysosomes are no longer viewed as organelles exclusively involved in catabolic pathways, but rather as highly dynamic elements of the autophagic-lysosomal pathway, involved in multiple cellular functions, including signaling, and able to adapt to environmental stimuli. This refined vision of lysosomes has substantially impacted on our understanding of the pathophysiology of lysosomal disorders. It is now clear that substrate accumulation triggers complex pathogenetic cascades that are responsible for disease pathology, such as aberrant vesicle trafficking, impairment of autophagy, dysregulation of signaling pathways, abnormalities of calcium homeostasis, and mitochondrial dysfunction. Novel technologies, in most cases based on high-throughput approaches, have significantly contributed to the characterization of lysosomal biology or lysosomal dysfunction and have the potential to facilitate diagnostic processes, and to enable the identification of new therapeutic targets.
溶酶体贮积症是一组由溶酶体功能的几种成分缺乏引起的代谢性疾病。最常受影响的是溶酶体水解酶,它们参与各种复杂分子和细胞结构的分解和再循环。随着时间的推移,对溶酶体生物学的理解逐渐提高。溶酶体不再被视为仅参与分解代谢途径的细胞器,而是作为自噬溶酶体途径的高度动态元件,参与多种细胞功能,包括信号转导,并能够适应环境刺激。这种对溶酶体的精细认识极大地影响了我们对溶酶体疾病病理生理学的理解。现在很清楚,底物积累会引发复杂的发病机制级联反应,导致疾病病理学,如异常囊泡运输、自噬受损、信号通路失调、钙稳态异常和线粒体功能障碍。新型技术,大多数情况下基于高通量方法,极大地促进了溶酶体生物学或溶酶体功能障碍的特征描述,并有可能促进诊断过程,并有助于确定新的治疗靶点。