Clinical Genetics Unit, Department of Pediatric Medicine, Giovanni XXIII Children's Hospital, Bari, Italy.
Department of Biomedical Science and Human Oncology, Pediatric Unit, University of Bari "Aldo Moro", Bari, Italy.
Mol Genet Genomic Med. 2020 Oct;8(10):e1371. doi: 10.1002/mgg3.1371. Epub 2020 Aug 11.
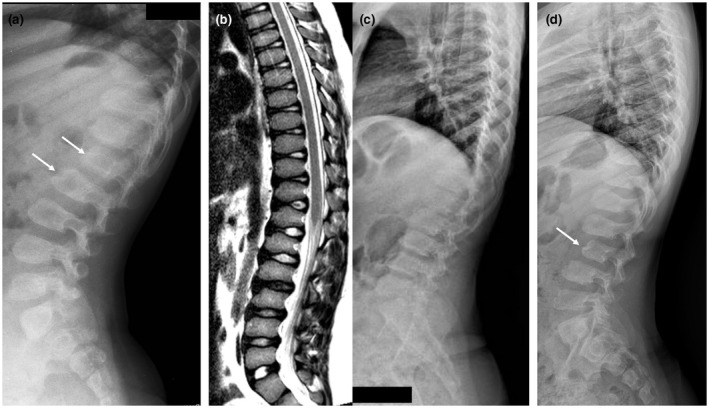
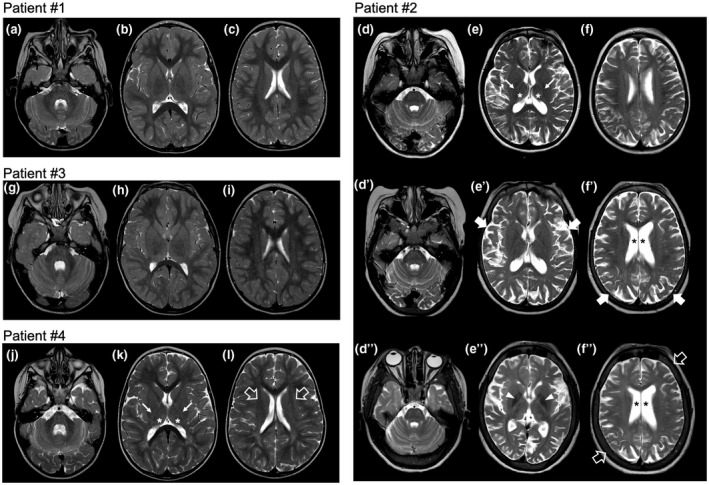
In GM1 gangliosidosis the lack of function of β-galactosidase results in an accumulation of GM1 ganglioside and related glycoconjugates in visceral organs, and particularly in the central nervous system, leading to severe disability and premature death. In the type 2 form of the disease, early intervention would be important to avoid precocious complications. To date, there are no effective therapeutic options in preventing progressive neurological deterioration. Substrate reduction therapy with Miglustat, a N-alkylated sugar that inhibits the enzyme glucosylceramide synthase, has been proposed for the treatment of several lysosomal storage disorders such as Gaucher type 1 and Niemann Pick Type C diseases. However, data on Miglustat therapy in patients with GM1 gangliosidosis are still scarce.
We report here the results of Miglustat administration in four Italian children (average age: 55 months, range 20-125) affected by GM1 gangliosidosis type 2 treated in three different Italian pediatric hospitals specialized in metabolic diseases.
This treatment was safe and relatively well tolerated by all patients, with stabilization and/or slowing down of the neurological progression in three subjects.
在 GM1 神经节苷脂贮积症中,β-半乳糖苷酶的缺乏导致 GM1 神经节苷脂和相关糖脂缀合物在内脏器官中积累,特别是在中枢神经系统中,导致严重的残疾和过早死亡。在疾病的 2 型中,早期干预对于避免过早出现并发症非常重要。迄今为止,还没有有效的治疗方法可以预防进行性神经恶化。用 N-烷基化糖麦格司他进行底物减少治疗,这种糖可以抑制葡萄糖脑苷脂合酶,已经被提议用于治疗几种溶酶体贮积症,如 Gaucher 1 型和 Niemann Pick C 型疾病。然而,关于麦格司他治疗 GM1 神经节苷脂贮积症患者的数据仍然很少。
我们在此报告了 Miglustat 在意大利的三个专门治疗代谢疾病的儿科医院中治疗的 4 名 GM1 神经节苷脂贮积症 2 型意大利儿童(平均年龄:55 个月,范围 20-125)中的结果。
这种治疗方法对所有患者均安全且相对耐受良好,其中 3 名患者的神经进展得到稳定和/或减缓。