Department of Systems Biology, Columbia University, New York, NY, USA.
Department of Applied Mathematics and Applied Physics, Columbia University, New York, NY, USA.
Nat Commun. 2021 Jan 21;12(1):510. doi: 10.1038/s41467-020-20847-0.
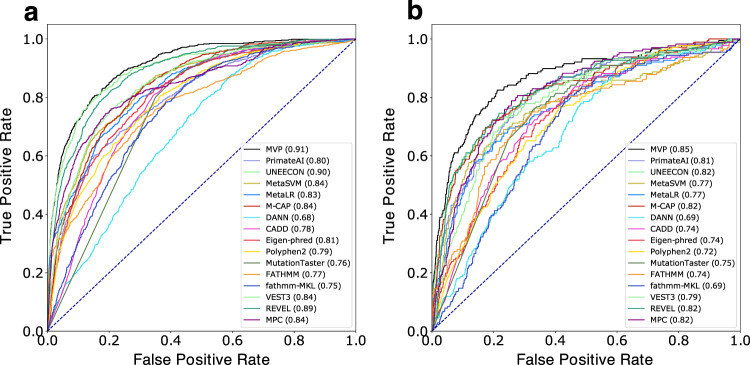
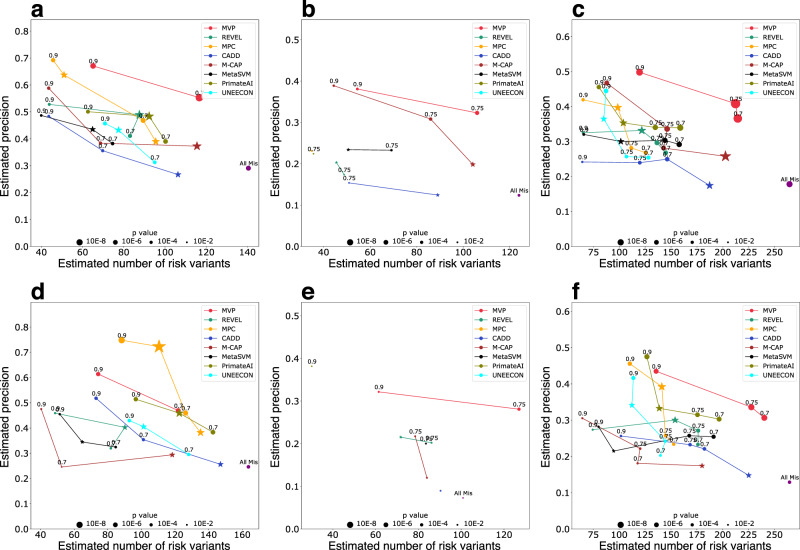
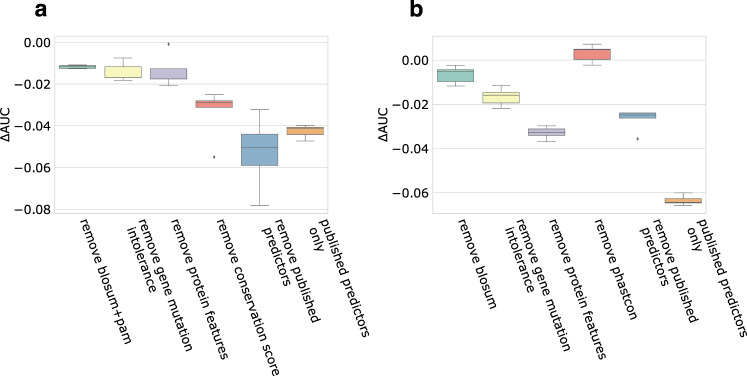
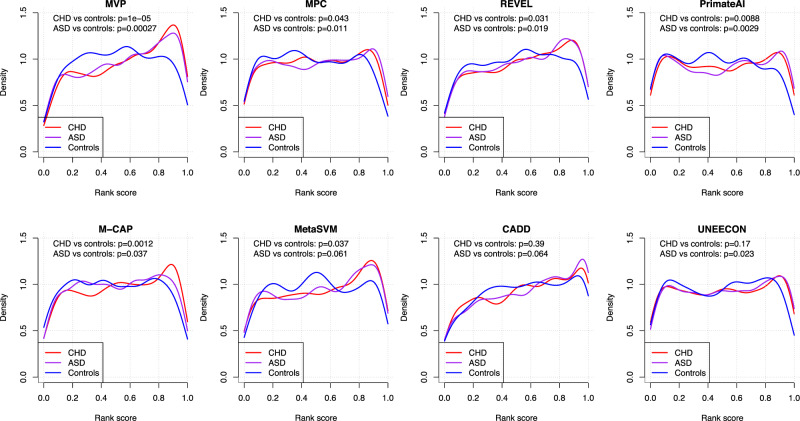
Accurate pathogenicity prediction of missense variants is critically important in genetic studies and clinical diagnosis. Previously published prediction methods have facilitated the interpretation of missense variants but have limited performance. Here, we describe MVP (Missense Variant Pathogenicity prediction), a new prediction method that uses deep residual network to leverage large training data sets and many correlated predictors. We train the model separately in genes that are intolerant of loss of function variants and the ones that are tolerant in order to take account of potentially different genetic effect size and mode of action. We compile cancer mutation hotspots and de novo variants from developmental disorders for benchmarking. Overall, MVP achieves better performance in prioritizing pathogenic missense variants than previous methods, especially in genes tolerant of loss of function variants. Finally, using MVP, we estimate that de novo coding variants contribute to 7.8% of isolated congenital heart disease, nearly doubling previous estimates.
准确预测错义变异的致病性在遗传研究和临床诊断中至关重要。先前发表的预测方法有助于错义变异的解读,但性能有限。在这里,我们描述了 MVP(错义变异致病性预测),这是一种新的预测方法,它使用深度残差网络来利用大型训练数据集和许多相关的预测因子。我们分别在功能丧失变异不易发生的基因和容忍的基因中训练模型,以考虑潜在的不同遗传效应大小和作用模式。我们编译癌症突变热点和发育障碍中的从头变异作为基准。总的来说,MVP 在优先考虑致病性错义变异方面的性能优于以前的方法,特别是在容忍功能丧失变异的基因中。最后,使用 MVP,我们估计新生编码变异导致 7.8%的孤立性先天性心脏病,几乎是之前估计的两倍。