Ophthalmology and Visual Sciences, Washington University School of Medicine, St. Louis, MO, USA.
FASEB J. 2021 Feb;35(2):e21288. doi: 10.1096/fj.202002037R.
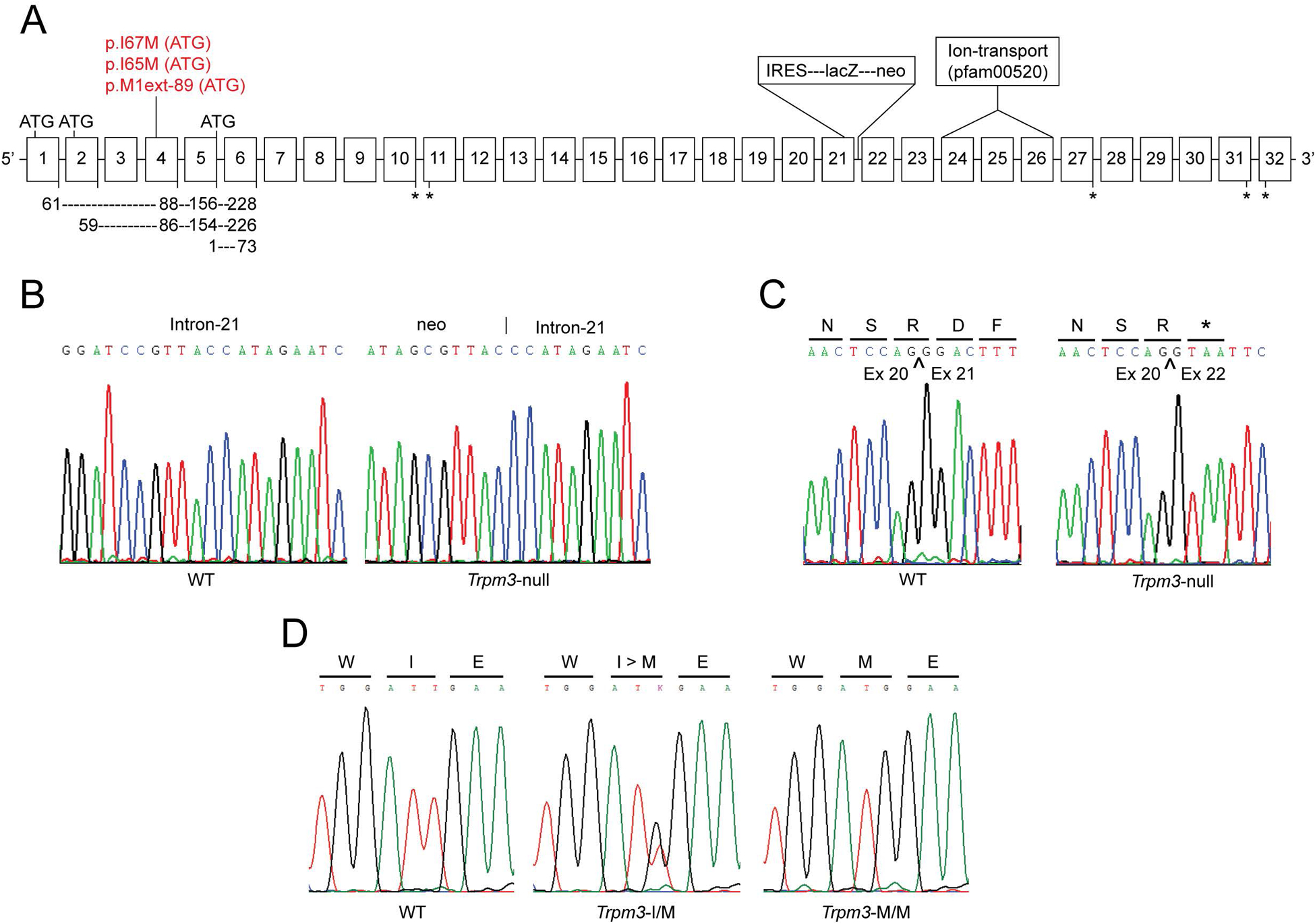
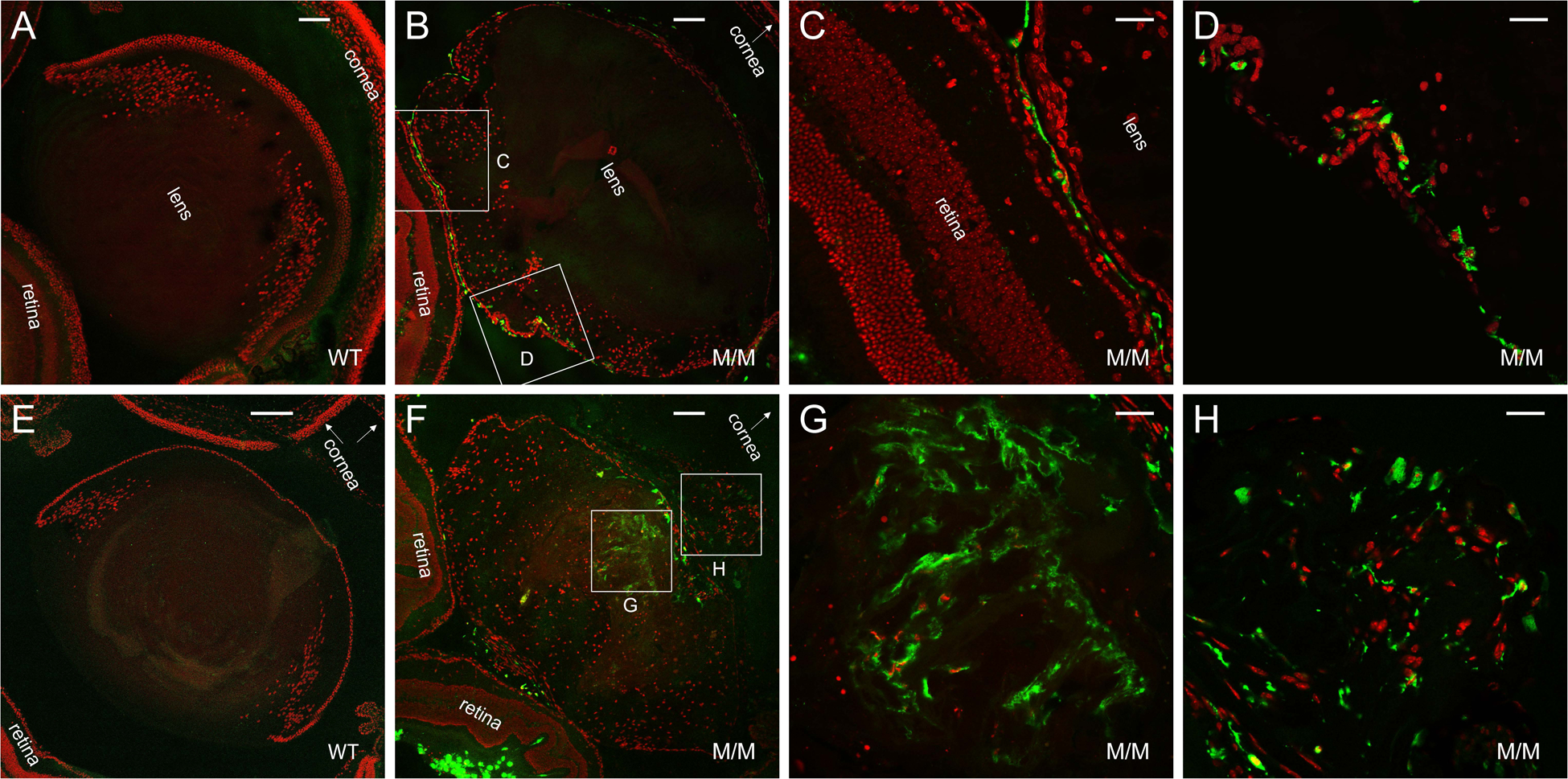
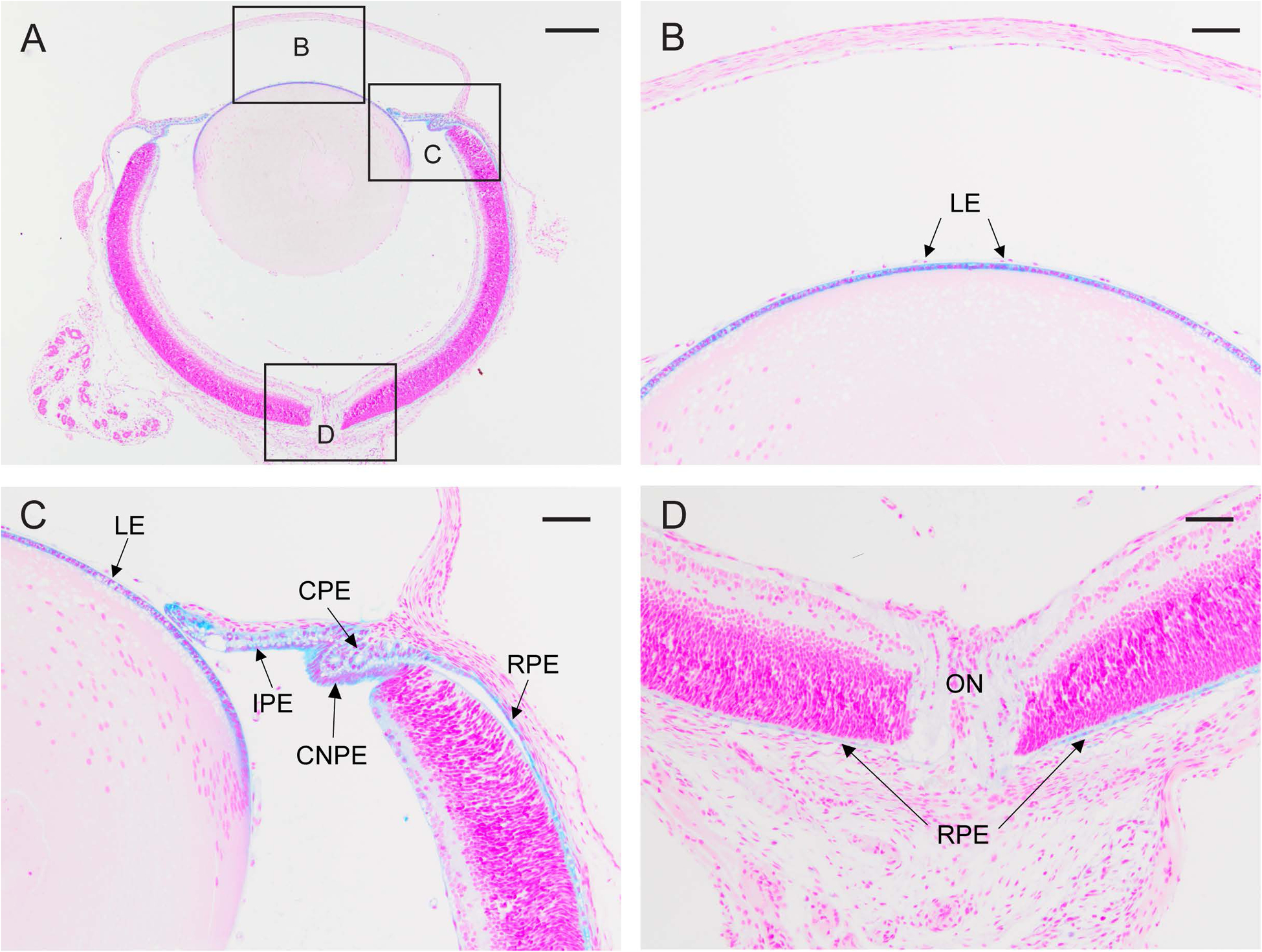
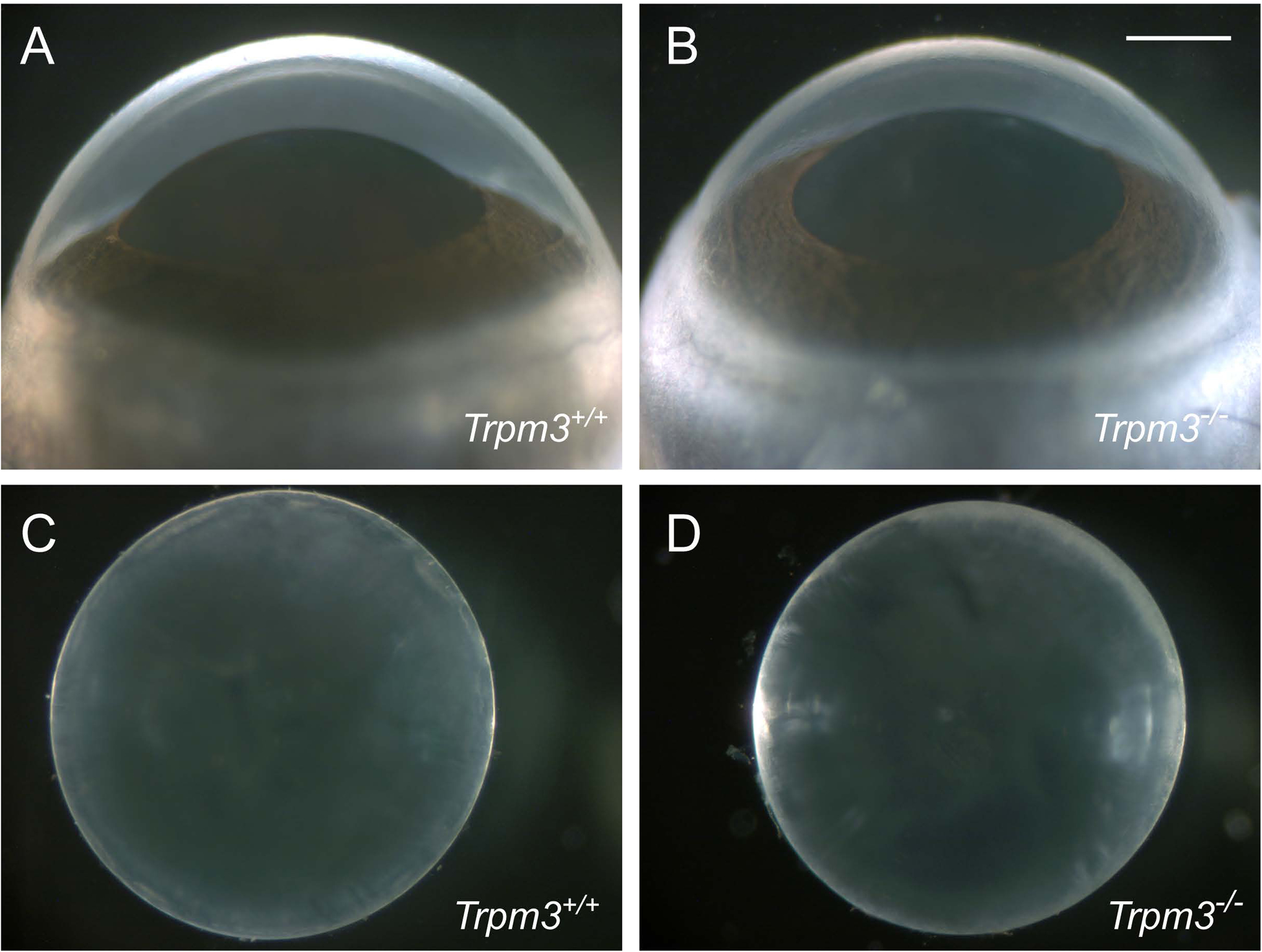
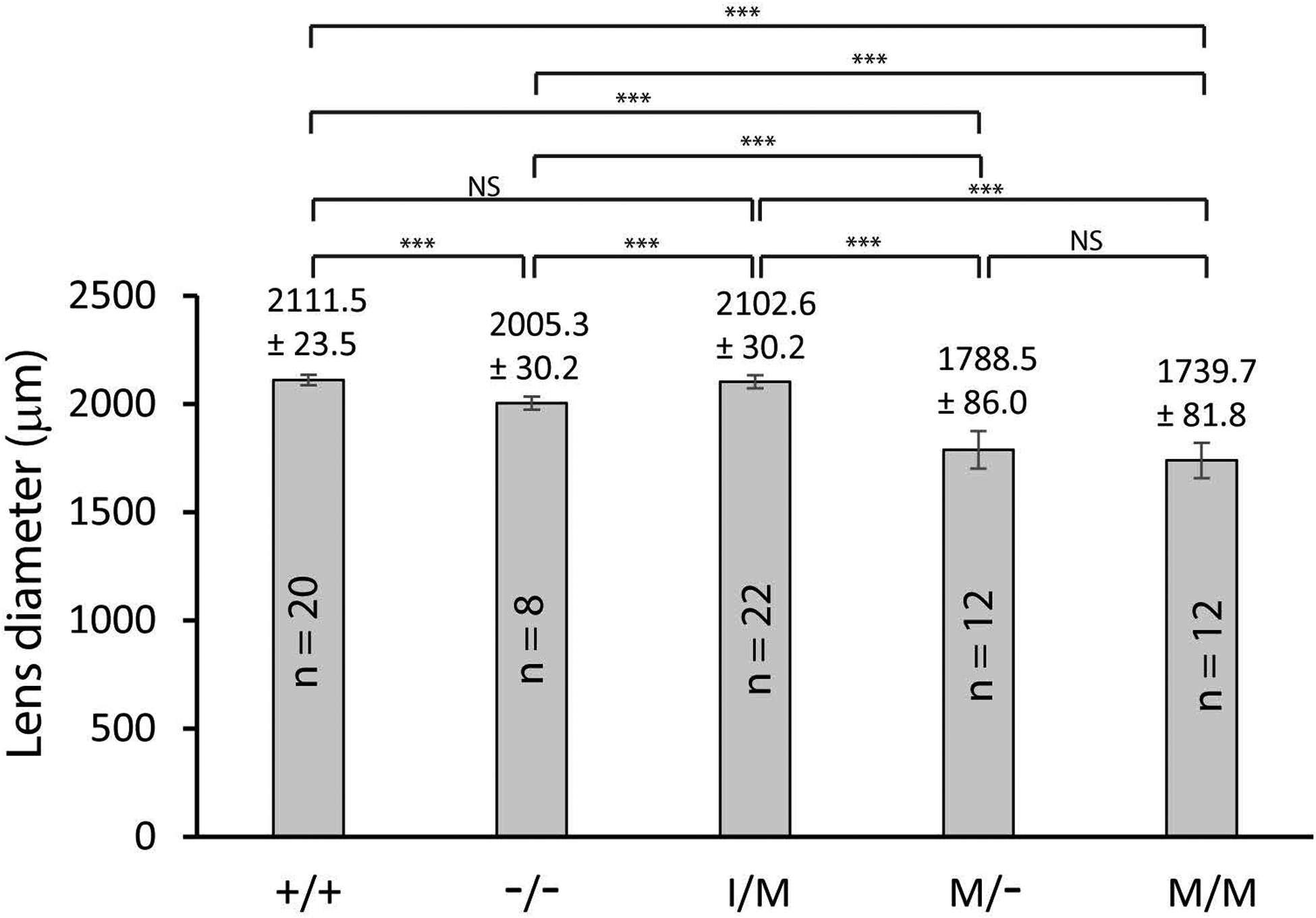
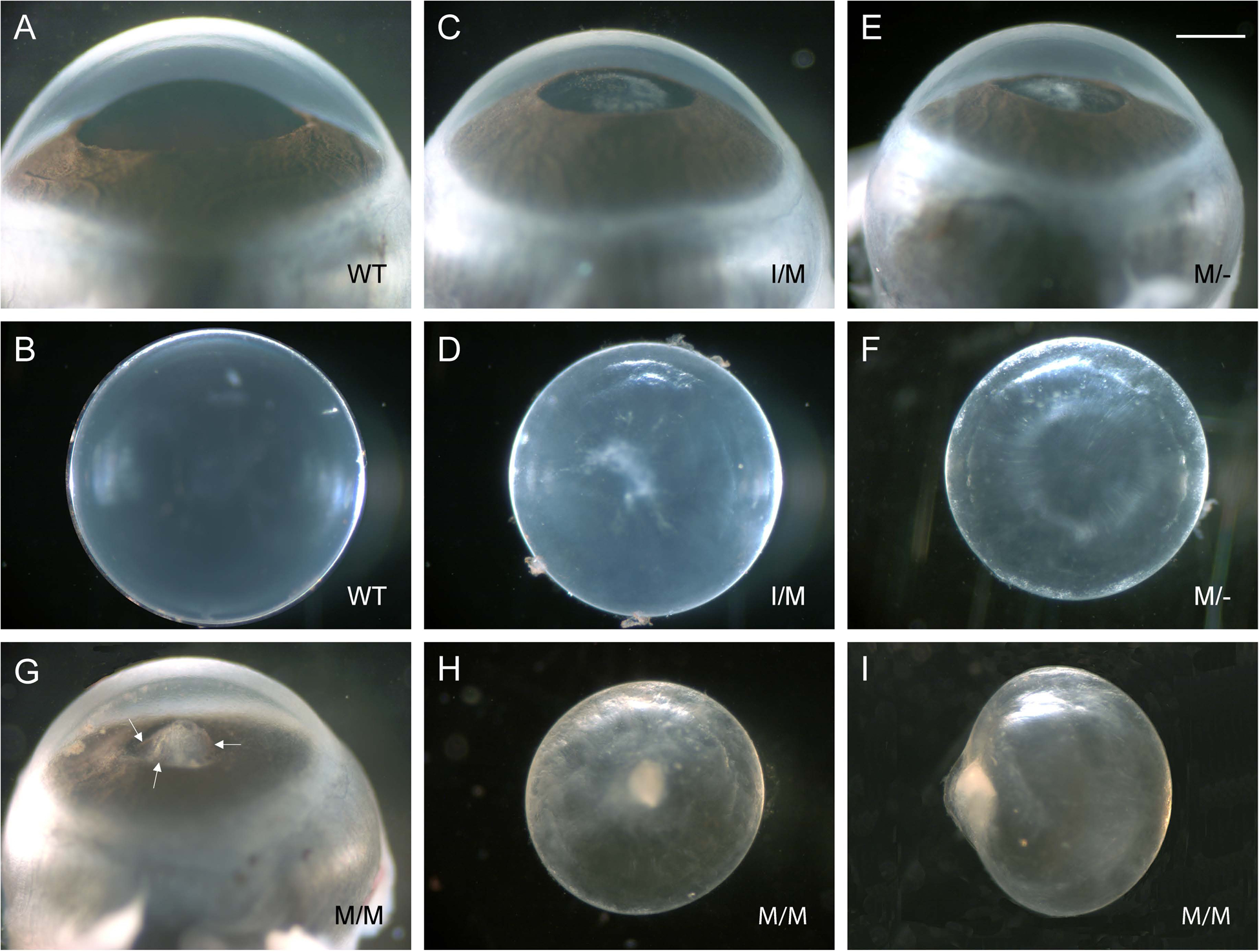
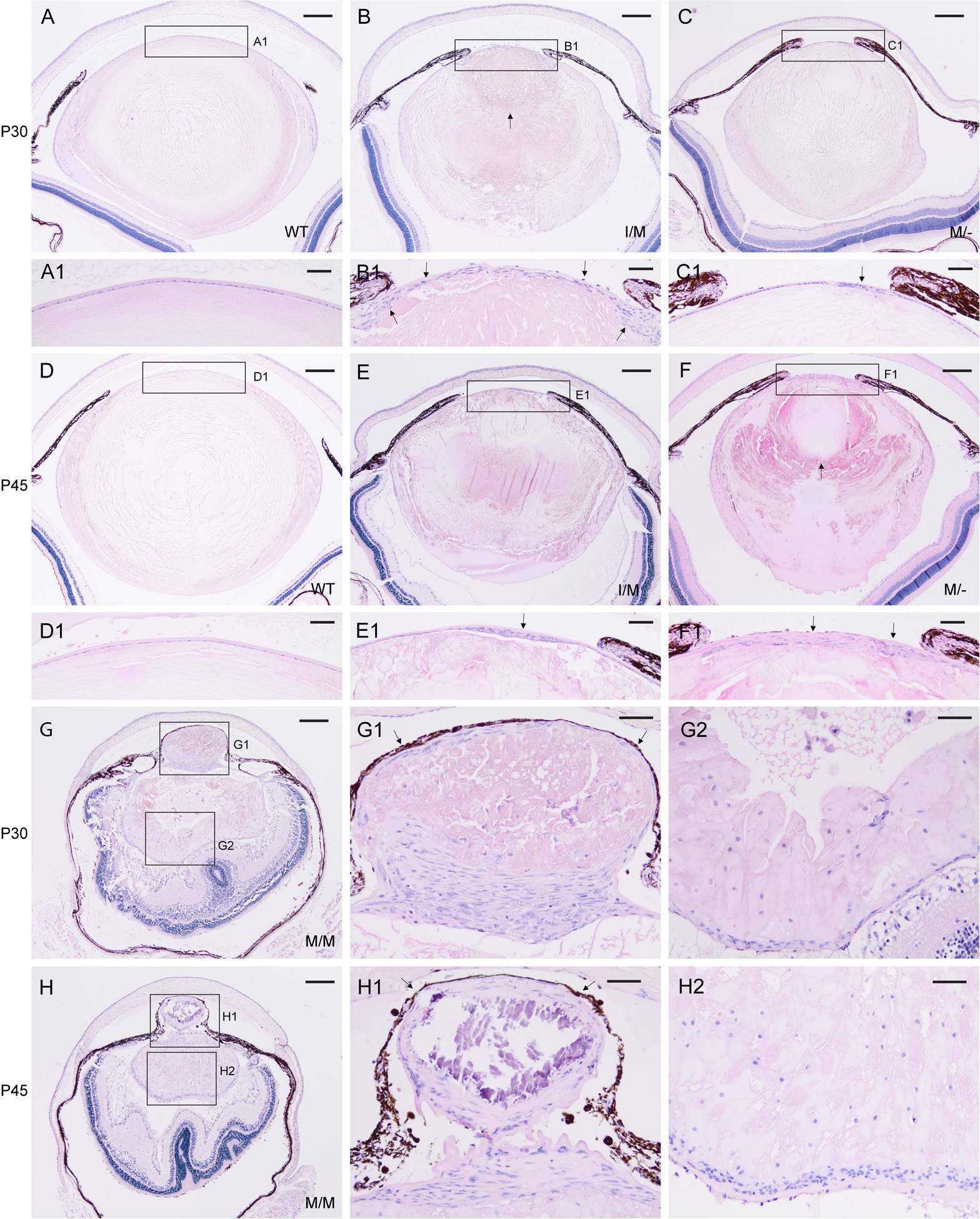
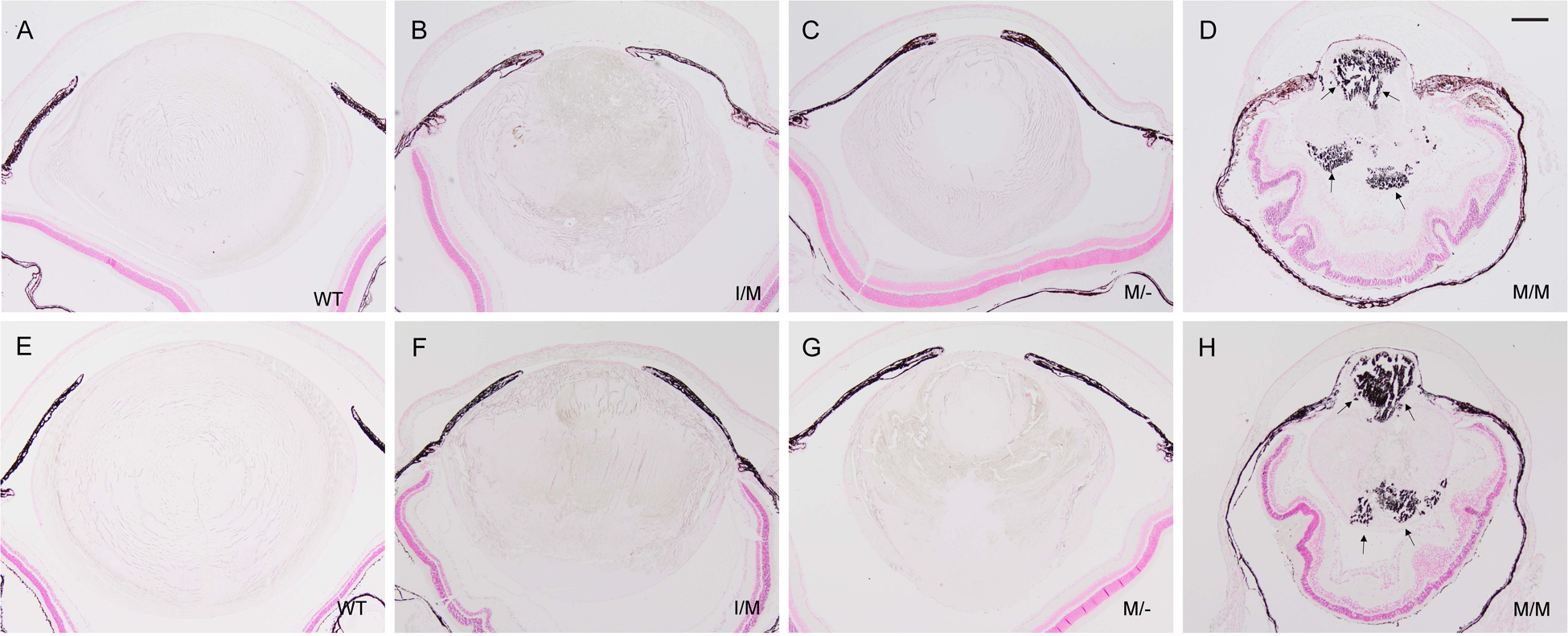
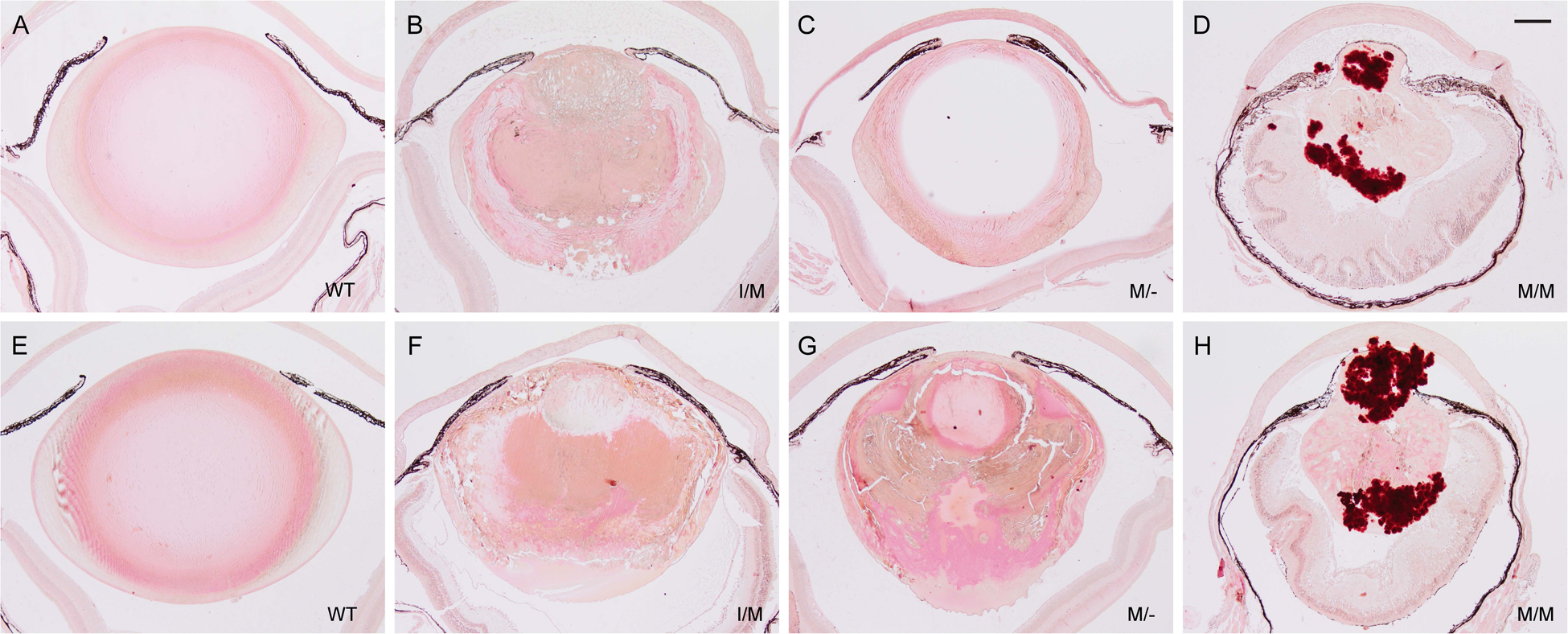
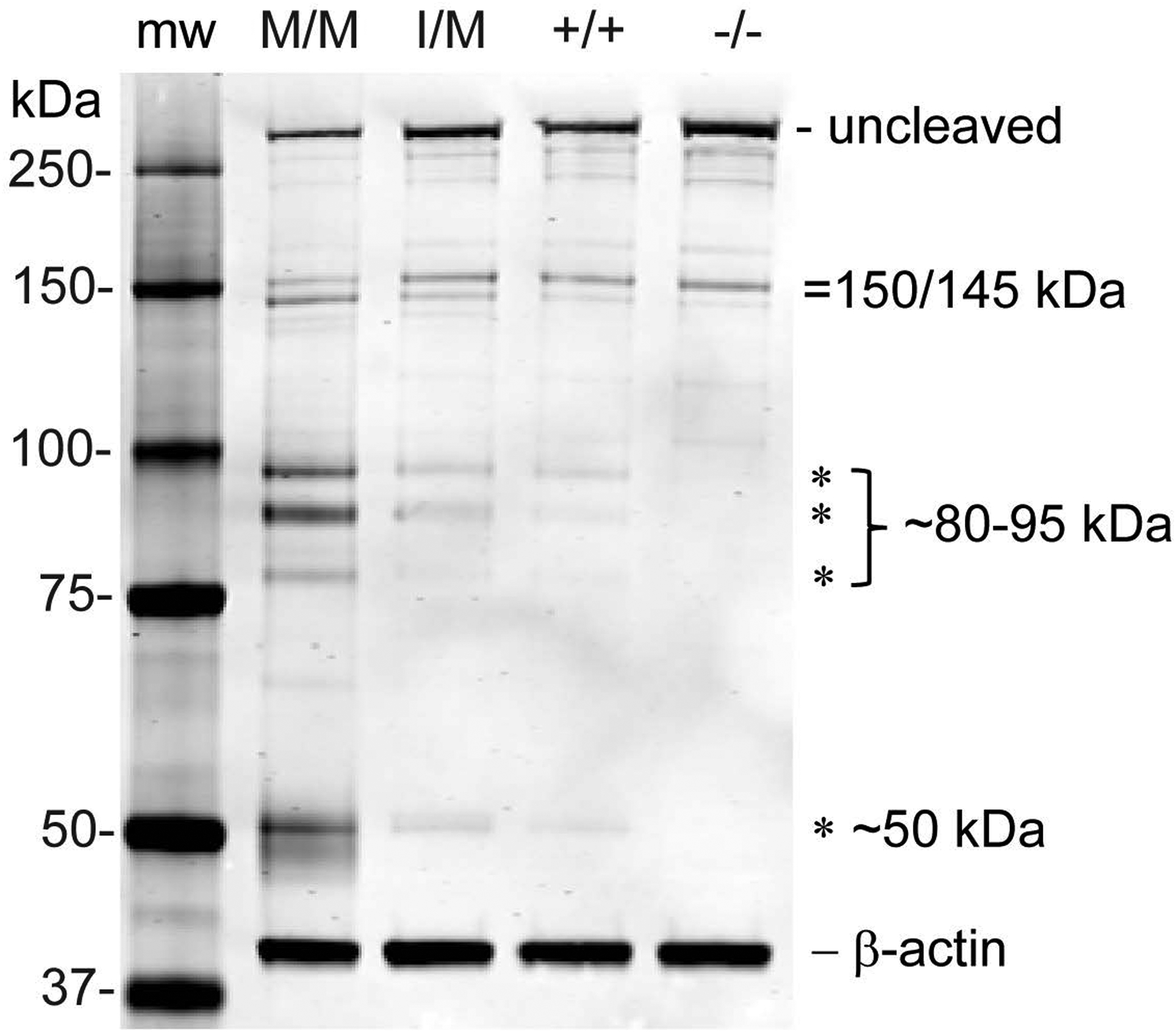
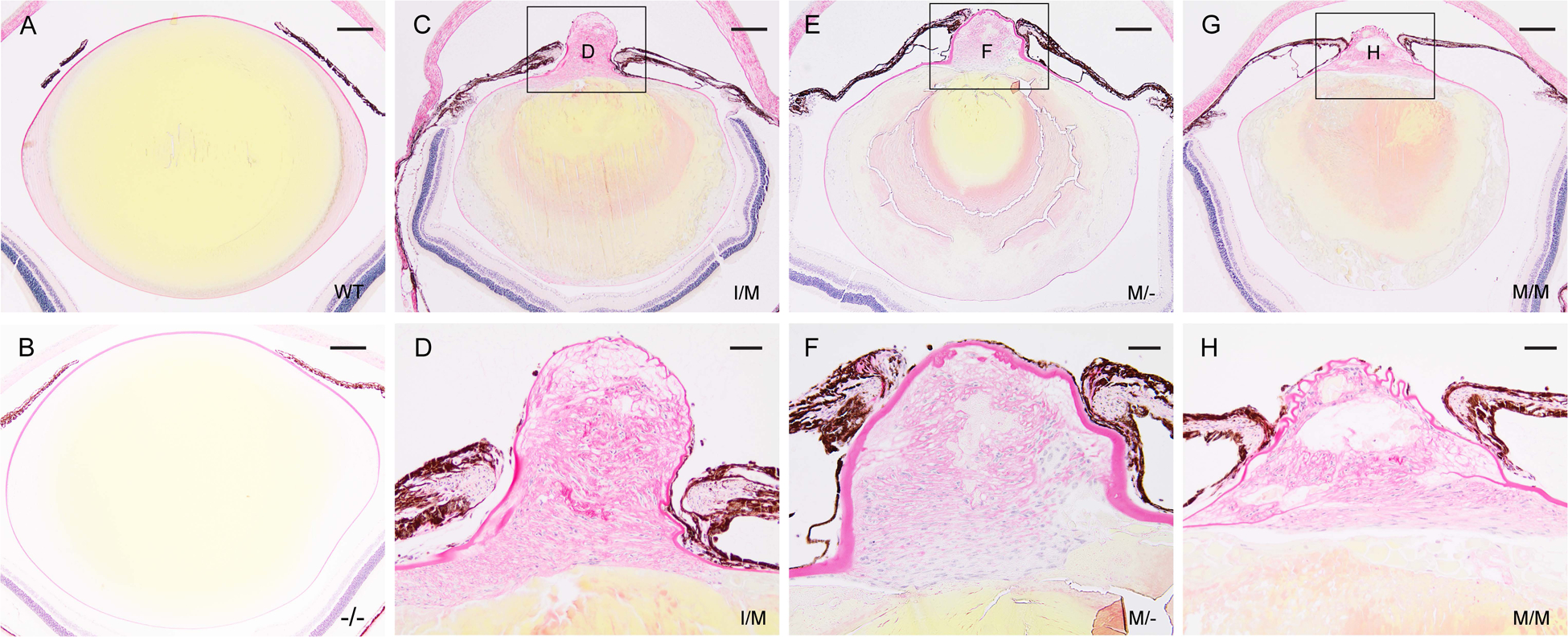
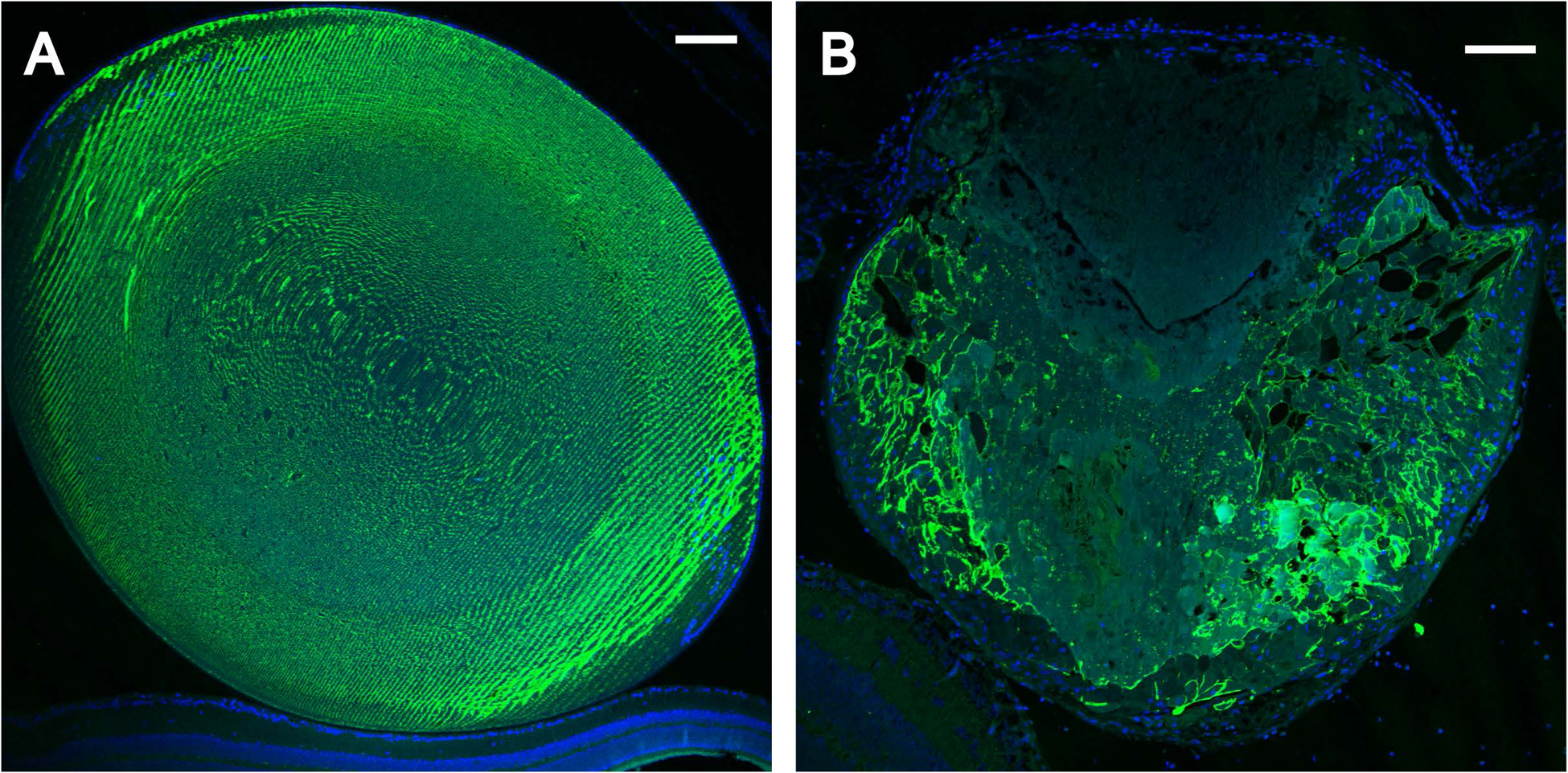
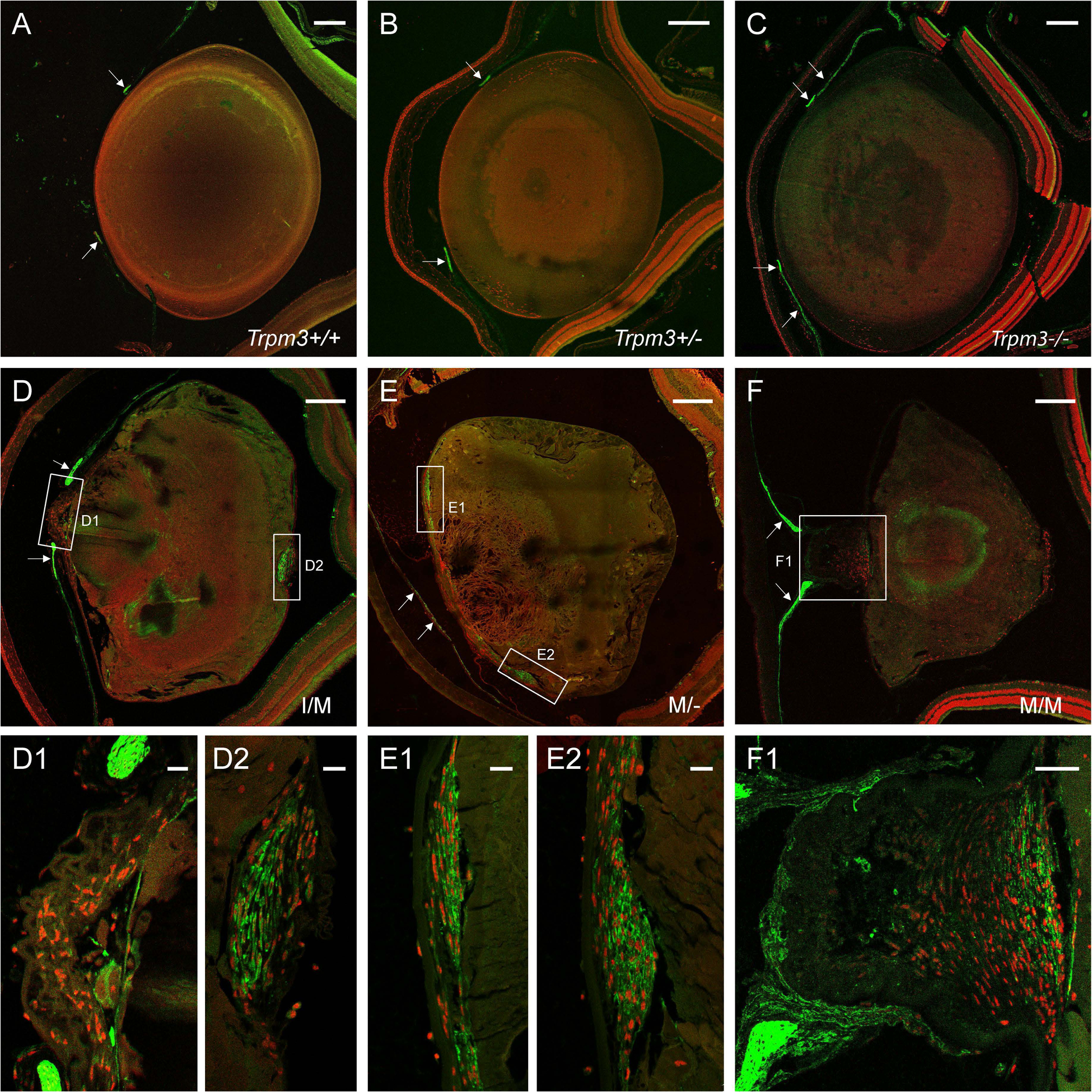
Transient-receptor-potential cation channel, subfamily M, member 3 (TRPM3) serves as a polymodal calcium sensor in diverse mammalian cell-types. Mutation of the human TRPM3 gene (TRPM3) has been linked with inherited forms of early-onset cataract with or without other eye abnormalities. Here, we have characterized the ocular phenotypes of germline "knock-in" mice that harbor a human cataract-associated isoleucine-to-methionine mutation (p.I65M) in TRPM3 (Trpm3-mutant) compared with germline "knock-out" mice that functionally lack TRPM3 (Trpm3-null). Despite strong expression of Trpm3 in lens epithelial cells, neither heterozygous (Trpm3 ) nor homozygous (Trpm3 ) Trpm3-null mice developed cataract; however, the latter exhibited a mild impairment of lens growth. In contrast, homozygous Trpm3-M/M mutants developed severe, progressive, anterior pyramid-like cataract with microphthalmia, whereas heterozygous Trpm3-I/M and hemizygous Trpm3-M/- mutants developed anterior pyramidal cataract with delayed onset and progression-consistent with a semi-dominant lens phenotype. Histochemical staining revealed abnormal accumulation of calcium phosphate-like deposits and collagen fibrils in Trpm3-mutant lenses and immunoblotting detected increased αII-spectrin cleavage products consistent with calpain hyper-activation. Immunofluorescent confocal microscopy of Trpm3-M/M mutant lenses revealed fiber cell membrane degeneration that was accompanied by accumulation of alpha-smooth muscle actin positive (α-SMA+ve) myofibroblast-like cells and macrosialin positive (CD68+ve) macrophage-like cells. Collectively, our mouse model data support an ocular disease association for TRPM3 in humans and suggest that (1) Trpm3 deficiency impaired lens growth but not lens transparency and (2) Trpm3 dysfunction resulted in progressive lens degeneration and calcification coupled with pro-fibrotic (α-SMA+ve) and immune (CD68+ve) cell responses.
瞬时受体电位阳离子通道亚家族 M 成员 3(TRPM3)作为一种多模式钙传感器,存在于多种哺乳动物细胞类型中。人类 TRPM3 基因(TRPM3)的突变与先天性早发性白内障有关,这些白内障伴有或不伴有其他眼部异常。在这里,我们对携带 TRPM3 中与白内障相关的异亮氨酸到蛋氨酸突变(p.I65M)的种系“敲入”(Trpm3-突变)小鼠与种系“敲除”(Trpm3-缺失)小鼠的眼部表型进行了特征描述,该突变导致 TRPM3 功能缺失。尽管 Trpm3 在晶状体上皮细胞中表达强烈,但杂合子(Trpm3 )和纯合子(Trpm3 )Trpm3-缺失小鼠均未发生白内障;然而,后者表现出晶状体生长的轻度受损。相比之下,纯合子 Trpm3-M/M 突变体发展为严重的、进行性的前金字塔状白内障伴小眼球,而杂合子 Trpm3-I/M 和半合子 Trpm3-M/-突变体发展为前金字塔状白内障,发病和进展延迟-与半显性晶状体表型一致。组织化学染色显示 Trpm3 突变体晶状体中异常积累钙磷样沉积物和胶原纤维,免疫印迹检测到钙蛋白酶过度激活导致αII- spectrin 裂解产物增加。Trpm3-M/M 突变体晶状体的免疫荧光共聚焦显微镜显示纤维细胞膜退化,伴有α-平滑肌肌动蛋白阳性(α-SMA+ve)肌成纤维细胞样细胞和巨唾液酸蛋白阳性(CD68+ve)巨噬细胞样细胞的积累。总的来说,我们的小鼠模型数据支持人类 TRPM3 与眼部疾病的关联,并表明(1)Trpm3 缺乏会损害晶状体生长,但不会损害晶状体透明度;(2)Trpm3 功能障碍导致进行性晶状体变性和钙化,伴有促纤维化(α-SMA+ve)和免疫(CD68+ve)细胞反应。