Institute of Pharmacy and Molecular Biotechnology, Im Neuenheimer Feld 364, 69120, Heidelberg, Germany.
Biochimie. 2021 Mar;182:177-184. doi: 10.1016/j.biochi.2021.01.010. Epub 2021 Jan 20.
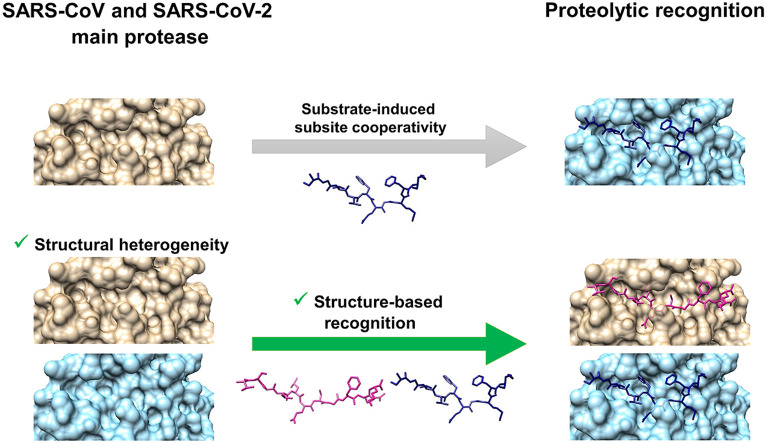
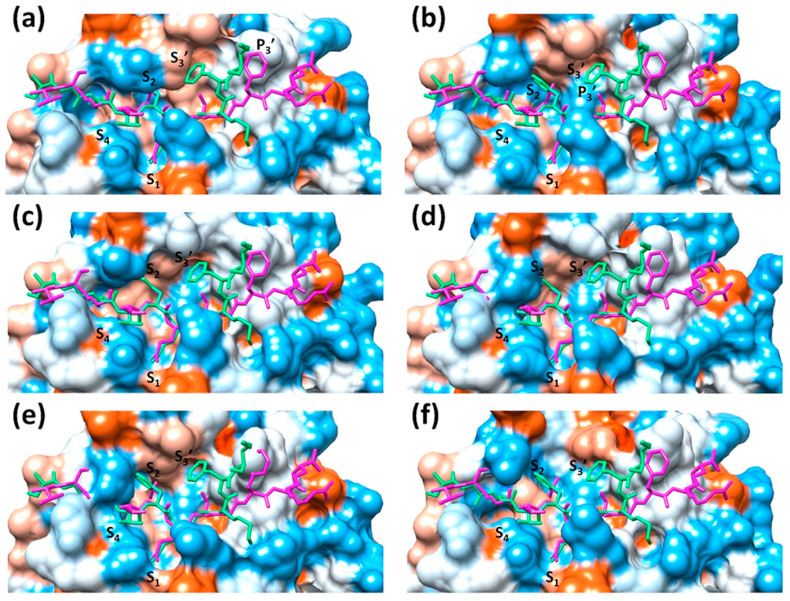
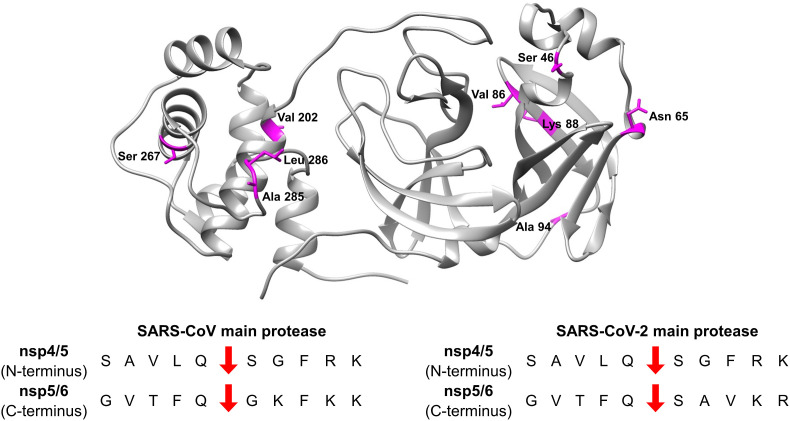
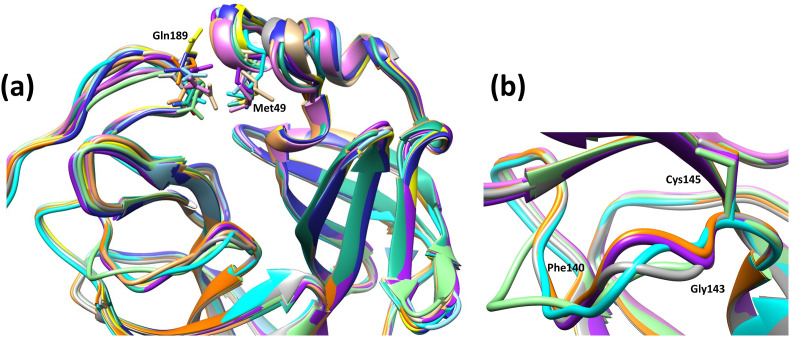
The main protease (M) of SARS-CoV and SARS-CoV-2 is a key enzyme in viral replication and a promising target for the development of antiviral therapeutics. The understanding of this protein is based on a number of observations derived from earlier x-ray structures, which mostly consider substrates or ligands as the main reason behind modulation of the active site. This lead to the concept of substrate-induced subsite cooperativity as an initial attempt to explain the dual binding specificity of this enzyme in recognizing the cleavage sequences at its N- and C-termini, which are important processing steps in obtaining the mature protease. The presented hypothesis proposes that structural heterogeneity is a property of the enzyme, independent of the presence of a substrate or ligand. Indeed, the analysis of M structures of SARS-CoV and SARS-CoV-2 reveals a conformational diversity for the catalytically competent state in ligand-free structures. Variation in the binding site appears to result from flexibility at residues lining the S subpocket and segments incorporating methionine 49 and glutamine 189. The structural evidence introduces "structure-based recognition" as a new paradigm in substrate proteolysis by M. In this concept, the binding space in subpockets of the enzyme varies in a non-cooperative manner, causing distinct conformations, which recognize and process different cleavage sites, as the N- and C-termini. Insights into the recognition basis of the protease provide explanation to the ordered processing of cleavage sites. The hypothesis expands the conformational space of the enzyme and consequently opportunities for drug development and repurposing efforts.
SARS-CoV 和 SARS-CoV-2 的主要蛋白酶(M)是病毒复制的关键酶,也是开发抗病毒治疗药物的有前途的靶点。对这种蛋白质的理解是基于许多早期 X 射线结构的观察结果,这些结果主要将底物或配体作为调节活性位点的主要原因。这导致了底物诱导亚基协同性的概念,作为解释该酶双结合特异性的初始尝试,以识别其 N 和 C 末端的切割序列,这是获得成熟蛋白酶的重要加工步骤。提出的假设表明,结构异质性是酶的固有特性,与底物或配体的存在无关。事实上,对 SARS-CoV 和 SARS-CoV-2 的 M 结构的分析揭示了在无配体结构中催化活性状态的构象多样性。结合位点的变化似乎是由于 S 亚口袋中残基的灵活性以及包含甲硫氨酸 49 和谷氨酰胺 189 的片段造成的。结构证据将“基于结构的识别”引入 M 的底物蛋白水解的新范例中。在这个概念中,酶的亚口袋中的结合空间以非协同的方式变化,导致不同的构象,这些构象识别和处理不同的切割位点,就像 N 和 C 末端一样。对蛋白酶识别基础的深入了解为切割位点的有序加工提供了解释。该假设扩展了酶的构象空间,从而为药物开发和重新利用提供了机会。