Mellis Anna-Theresa, Roeper Juliane, Misko Albert L, Kohl Joshua, Schwarz Guenter
Department of Chemistry, Institute for Biochemistry, University of Cologne, Cologne, Germany.
Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States.
Front Genet. 2021 Jan 7;11:594828. doi: 10.3389/fgene.2020.594828. eCollection 2020.
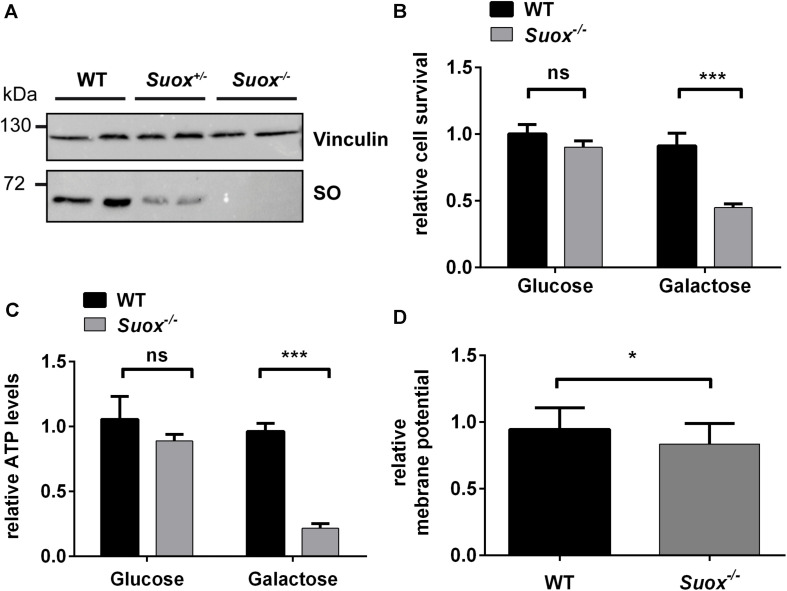
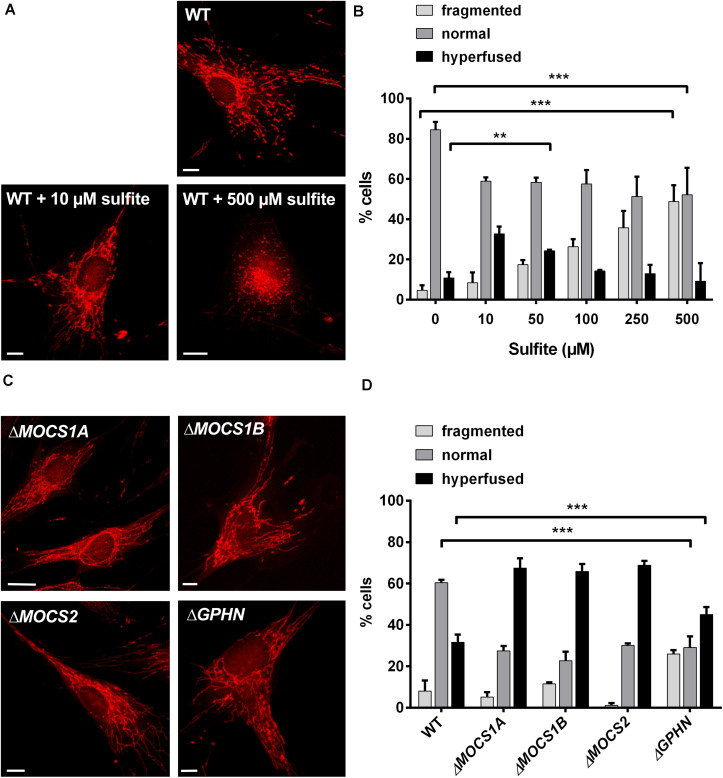
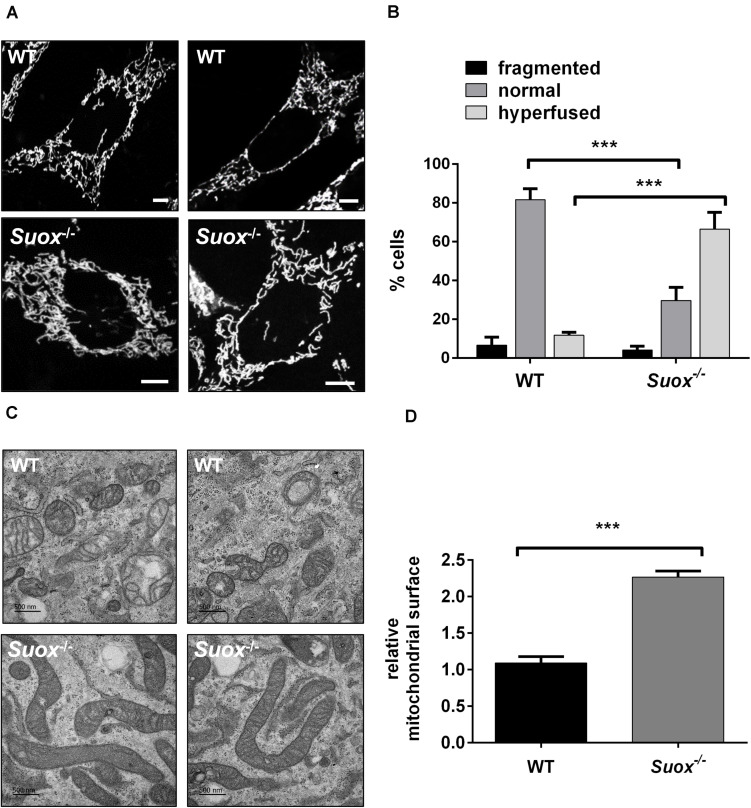
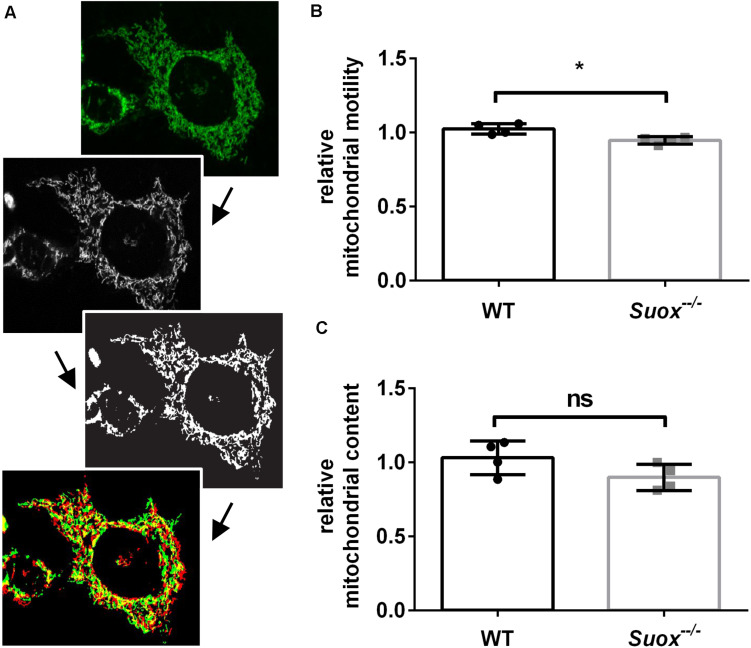
Molybdenum cofactor deficiency (MoCD) is an autosomal recessive disorder belonging to the large family of inborn errors in metabolism. Patients typically present with encephalopathy and seizures early after birth and develop severe neurodegeneration within the first few weeks of life. The main pathomechanism underlying MoCD is the loss of function of sulfite oxidase (SO), a molybdenum cofactor (Moco) dependent enzyme located in mitochondrial intermembrane space. SO catalyzes the oxidation of sulfite (SO ) to sulfate (SO ) in the terminal reaction of cysteine catabolism, and in the absence of its activity, sulfurous compounds such as SO , S-sulfocysteine, and thiosulfate accumulate in patients. Despite growing evidence that these compounds affect neuronal and mitochondrial function, the molecular basis of neuronal dysfunction and cell death in MoCD is still poorly understood. Here we show that mitochondria are severely affected by the loss of SO activity. SO-deficient mouse embryonic fibroblasts display reduced growth rates and impaired ATP production when cultured in galactose, which is an indicator of mitochondrial dysfunction. We also found that mitochondria in SO-deficient cells form a highly interconnected network compared to controls while displaying a slight decrease in motility and unchanged mitochondrial mass. Moreover, we show that the mitochondrial network is directly influenced by SO , as a moderate elevation of SO lead to the formation of an interconnected mitochondrial network, while high SO levels induced fragmentation. Finally, we found a highly interconnected mitochondrial network in MoCD patient-derived fibroblasts, similar to our findings in mouse-derived fibroblasts. We therefore conclude that altered mitochondrial dynamics are an important contributor to the disease phenotype and suggest that MoCD should be included among the mitochondrial disorders.
钼辅因子缺乏症(MoCD)是一种常染色体隐性疾病,属于先天性代谢缺陷大家族。患者通常在出生后早期出现脑病和癫痫发作,并在出生后的头几周内发生严重的神经退行性变。MoCD的主要发病机制是亚硫酸盐氧化酶(SO)功能丧失,SO是一种位于线粒体内膜间隙的钼辅因子(Moco)依赖性酶。SO在半胱氨酸分解代谢的终末反应中催化亚硫酸盐(SO₃²⁻)氧化为硫酸盐(SO₄²⁻),在其活性缺乏时,亚硫酸盐、S-磺基半胱氨酸和硫代硫酸盐等含硫化合物会在患者体内蓄积。尽管越来越多的证据表明这些化合物会影响神经元和线粒体功能,但MoCD中神经元功能障碍和细胞死亡的分子基础仍知之甚少。在此我们表明,线粒体受到SO活性丧失的严重影响。缺乏SO的小鼠胚胎成纤维细胞在以半乳糖培养时生长速率降低且ATP生成受损,这是线粒体功能障碍的一个指标。我们还发现,与对照组相比,缺乏SO的细胞中的线粒体形成高度相互连接的网络,同时运动性略有降低且线粒体质量不变。此外,我们表明线粒体网络直接受SO₃²⁻影响,因为适度升高SO₃²⁻会导致形成相互连接的线粒体网络,而高SO₃²⁻水平会诱导碎片化。最后,我们在MoCD患者来源的成纤维细胞中发现了高度相互连接的线粒体网络,类似于我们在小鼠来源的成纤维细胞中的发现。因此,我们得出结论,线粒体动力学改变是疾病表型的一个重要促成因素,并建议MoCD应纳入线粒体疾病范畴。