Department of Theory and Bio-Systems, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany.
NMR Group-Institute for Physics, Martin-Luther University Halle-Wittenberg, 06120 Halle (Saale), Germany.
J Chem Inf Model. 2021 Feb 22;61(2):938-949. doi: 10.1021/acs.jcim.0c01299. Epub 2021 Jan 26.
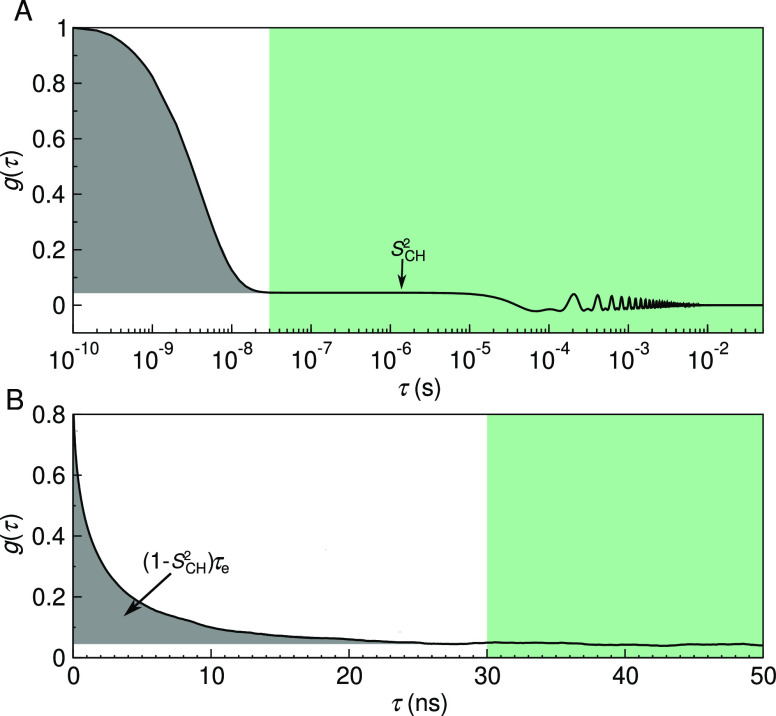
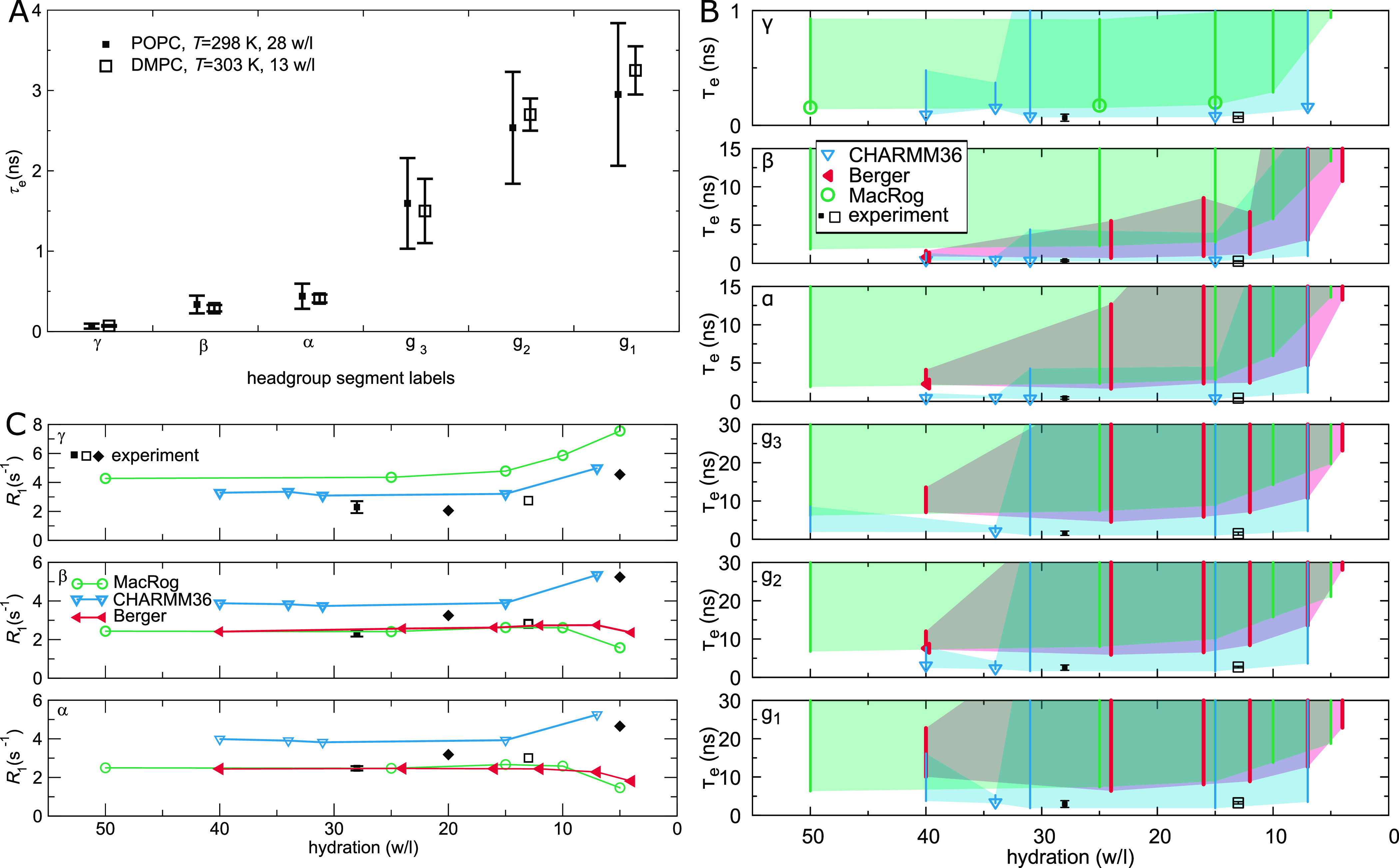
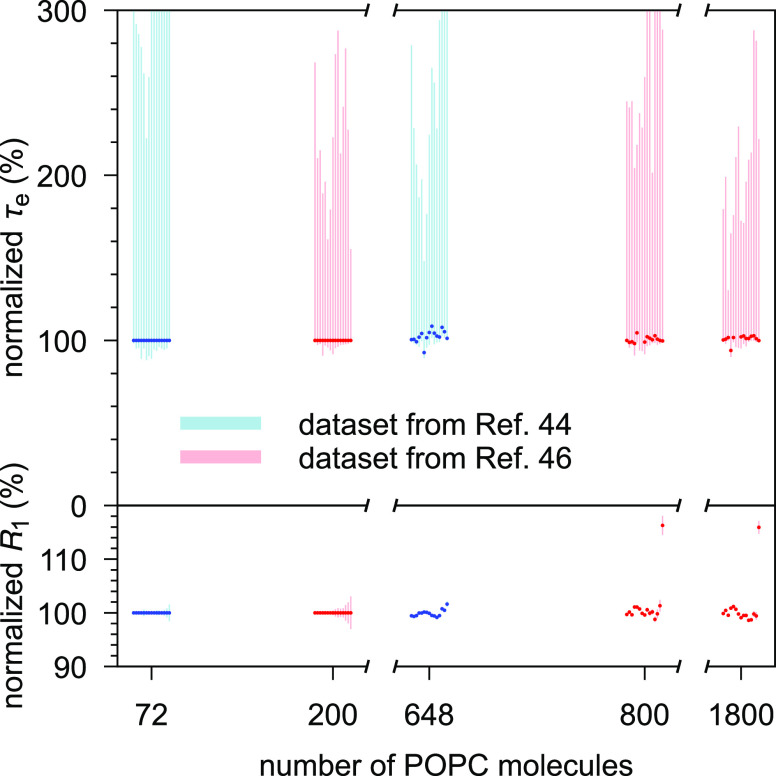
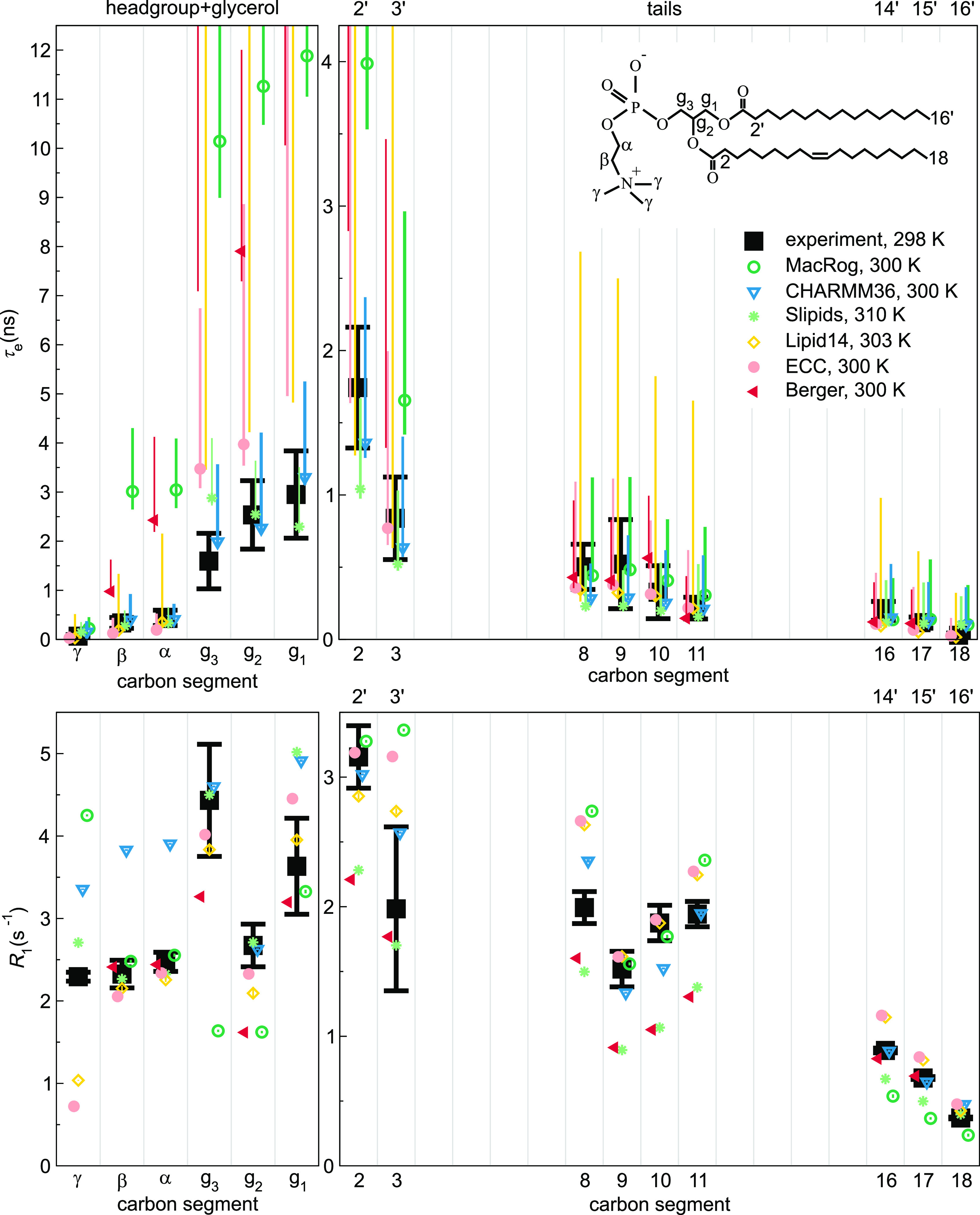
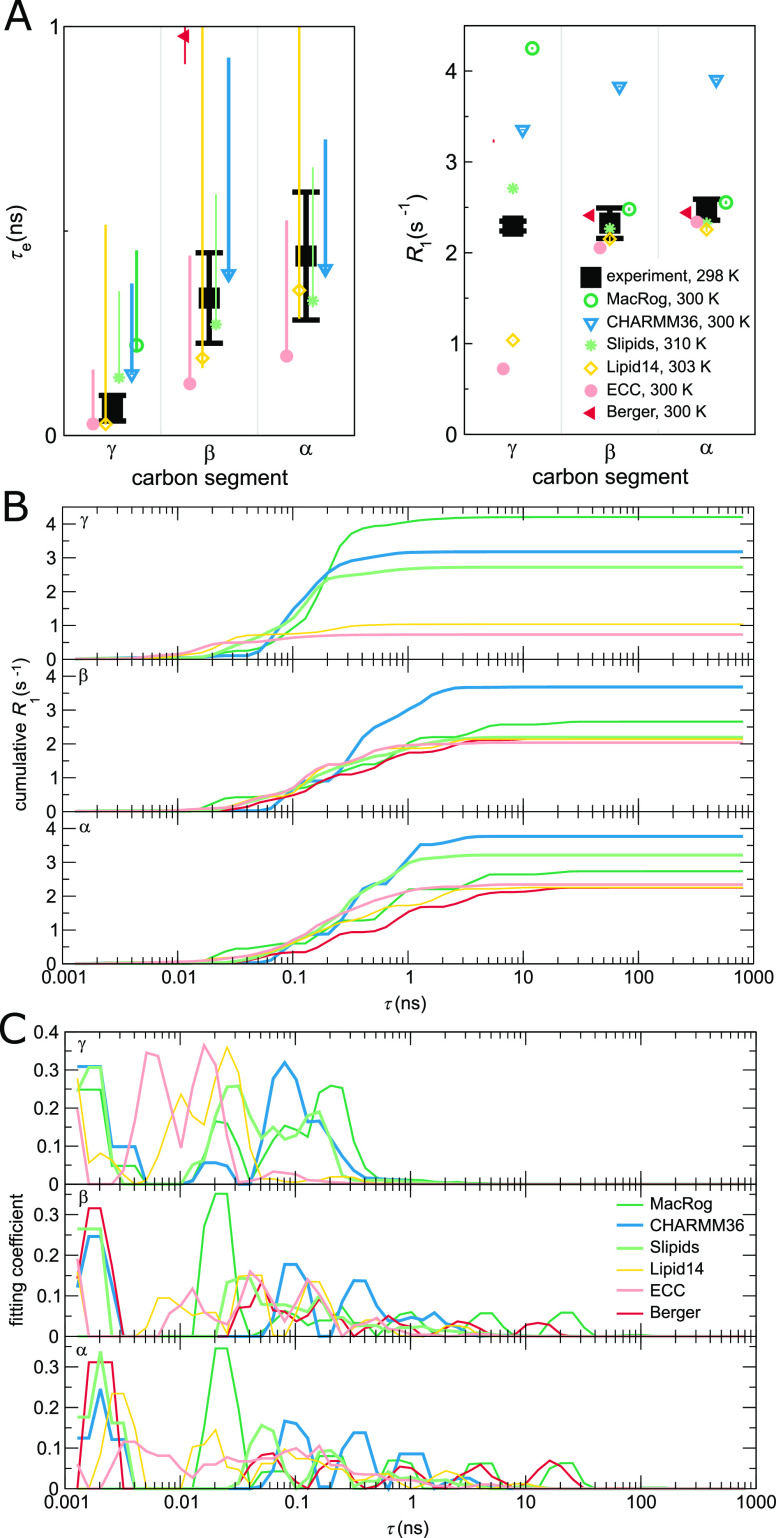
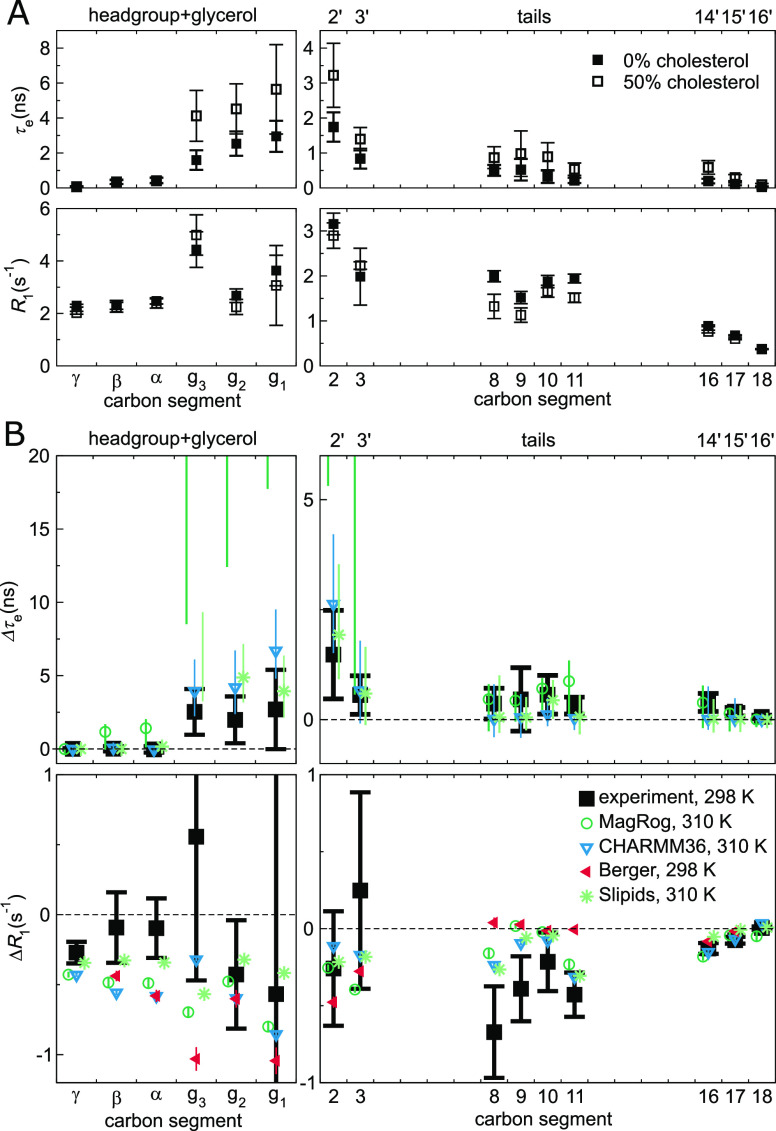
Molecular dynamics (MD) simulations are widely used to monitor time-resolved motions of biomacromolecules, although it often remains unknown how closely the conformational dynamics correspond to those occurring in real life. Here, we used a large set of open-access MD trajectories of phosphatidylcholine (PC) lipid bilayers to benchmark the conformational dynamics in several contemporary MD models (force fields) against nuclear magnetic resonance (NMR) data available in the literature: effective correlation times and spin-lattice relaxation rates. We found none of the tested MD models to fully reproduce the conformational dynamics. That said, the dynamics in CHARMM36 and Slipids are more realistic than in the Amber Lipid14, OPLS-based MacRog, and GROMOS-based Berger force fields, whose sampling of the glycerol backbone conformations is too slow. The performance of CHARMM36 persists when cholesterol is added to the bilayer, and when the hydration level is reduced. However, for conformational dynamics of the PC headgroup, both with and without cholesterol, Slipids provides the most realistic description because CHARMM36 overestimates the relative weight of ∼1 ns processes in the headgroup dynamics. We stress that not a single new simulation was run for the present work. This demonstrates the worth of open-access MD trajectory databanks for the indispensable step of any serious MD study: benchmarking the available force fields. We believe this proof of principle will inspire other novel applications of MD trajectory databanks and thus aid in developing biomolecular MD simulations into a true computational microscope-not only for lipid membranes but for all biomacromolecular systems.
分子动力学(MD)模拟被广泛用于监测生物大分子的时间分辨运动,尽管通常仍不清楚构象动力学与实际发生的动力学之间的吻合程度。在这里,我们使用了一组大型的公开可用的 MD 轨迹来对磷脂酰胆碱(PC)脂质双层中的构象动力学进行基准测试,以评估几种当代 MD 模型(力场)与文献中可用的核磁共振(NMR)数据的吻合程度:有效相关时间和自旋晶格弛豫率。我们发现没有一个测试的 MD 模型能够完全再现构象动力学。也就是说,CHARMM36 和 Slipids 的动力学比 Amber Lipid14、基于 OPLS 的 MacRog 和基于 GROMOS 的 Berger 力场更现实,后两者甘油骨架构象的采样速度太慢。当向双层添加胆固醇或降低水合水平时,CHARMM36 的性能仍然存在。然而,对于 PC 头基的构象动力学,无论是有胆固醇还是没有胆固醇,Slipids 都提供了最现实的描述,因为 CHARMM36 高估了头基动力学中约 1ns 过程的相对权重。我们强调,为本研究没有运行新的模拟。这证明了公开可用的 MD 轨迹数据库对于任何严肃的 MD 研究的不可或缺的步骤的价值:基准测试可用的力场。我们相信,这一原理验证将激发 MD 轨迹数据库的其他新应用,从而有助于将生物分子 MD 模拟发展成为一种真正的计算显微镜——不仅适用于脂质膜,也适用于所有生物大分子系统。