Cardiovascular Institute, Department of Medicine, Allegheny Health Network, Pittsburgh, PA.
Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine, Department of Medicine, Emory University School of Medicine, Atlanta, GA.
Blood Adv. 2021 Jan 26;5(2):399-413. doi: 10.1182/bloodadvances.2020002754.
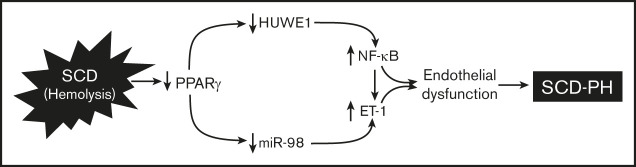
Sickle cell disease (SCD)-associated pulmonary hypertension (PH) causes significant morbidity and mortality. Here, we defined the role of endothelial specific peroxisome proliferator-activated receptor γ (PPARγ) function and novel PPARγ/HUWE1/miR-98 signaling pathways in the pathogenesis of SCD-PH. PH and right ventricular hypertrophy (RVH) were increased in chimeric Townes humanized sickle cell (SS) mice with endothelial-targeted PPARγ knockout (SSePPARγKO) compared with chimeric littermate control (SSLitCon). Lung levels of PPARγ, HUWE1, and miR-98 were reduced in SSePPARγKO mice compared with SSLitCon mice, whereas SSePPARγKO lungs were characterized by increased levels of p65, ET-1, and VCAM1. Collectively, these findings indicate that loss of endothelial PPARγ is sufficient to increase ET-1 and VCAM1 that contribute to endothelial dysfunction and SCD-PH pathogenesis. Levels of HUWE1 and miR-98 were decreased, and p65 levels were increased in the lungs of SS mice in vivo and in hemin-treated human pulmonary artery endothelial cells (HPAECs) in vitro. Although silencing of p65 does not regulate HUWE1 levels, the loss of HUWE1 increased p65 levels in HPAECs. Overexpression of PPARγ attenuated hemin-induced reductions of HUWE1 and miR-98 and increases in p65 and endothelial dysfunction. Similarly, PPARγ activation attenuated baseline PH and RVH and increased HUWE1 and miR-98 in SS lungs. In vitro, hemin treatment reduced PPARγ, HUWE1, and miR-98 levels and increased p65 expression, HPAEC monocyte adhesion, and proliferation. These derangements were attenuated by pharmacological PPARγ activation. Targeting these signaling pathways can favorably modulate a spectrum of pathobiological responses in SCD-PH pathogenesis, highlighting novel therapeutic targets in SCD pulmonary vascular dysfunction and PH.
镰状细胞病(SCD)相关肺动脉高压(PH)导致显著的发病率和死亡率。在这里,我们定义了内皮特异性过氧化物酶体增殖物激活受体γ(PPARγ)功能和新型 PPARγ/HUWE1/miR-98 信号通路在 SCD-PH 发病机制中的作用。与嵌合同胞对照(SSLitCon)相比,内皮靶向 PPARγ 敲除(SSePPARγKO)的嵌合 Townes 人源化镰状细胞(SS)小鼠中 PH 和右心室肥厚(RVH)增加。与 SSLitCon 小鼠相比,SSePPARγKO 小鼠的肺组织中 PPARγ、HUWE1 和 miR-98 水平降低,而 SSePPARγKO 肺组织的特征是 p65、ET-1 和 VCAM1 水平增加。总的来说,这些发现表明内皮 PPARγ 的缺失足以增加 ET-1 和 VCAM1,从而导致内皮功能障碍和 SCD-PH 发病机制。体内 SS 小鼠和体外血红素处理的人肺动脉内皮细胞(HPAECs)中,HUWE1 和 miR-98 的水平降低,p65 水平升高。虽然沉默 p65 不能调节 HUWE1 水平,但 HUWE1 的缺失增加了 HPAECs 中的 p65 水平。PPARγ 的过表达减弱了血红素诱导的 HUWE1 和 miR-98 的减少以及 p65 和内皮功能障碍的增加。同样,PPARγ 激活减轻了 SS 肺中的基础 PH 和 RVH 并增加了 HUWE1 和 miR-98。在体外,血红素处理降低了 PPARγ、HUWE1 和 miR-98 的水平并增加了 p65 的表达、HPAEC 单核细胞黏附和增殖。这些紊乱通过药理学 PPARγ 激活得到缓解。靶向这些信号通路可以有利地调节 SCD-PH 发病机制中一系列病理生物学反应,强调了 SCD 肺血管功能障碍和 PH 的新治疗靶点。