Institute of Human Genetics, Julius Maximilians University Würzburg, Würzburg, Germany.
Department of Otolaryngology-Head and Neck Surgery, Tübingen Hearing Research Centre, Eberhard Karls University Tübingen, Tübingen, Germany.
Hum Genet. 2021 Jun;140(6):915-931. doi: 10.1007/s00439-020-02254-z. Epub 2021 Jan 26.
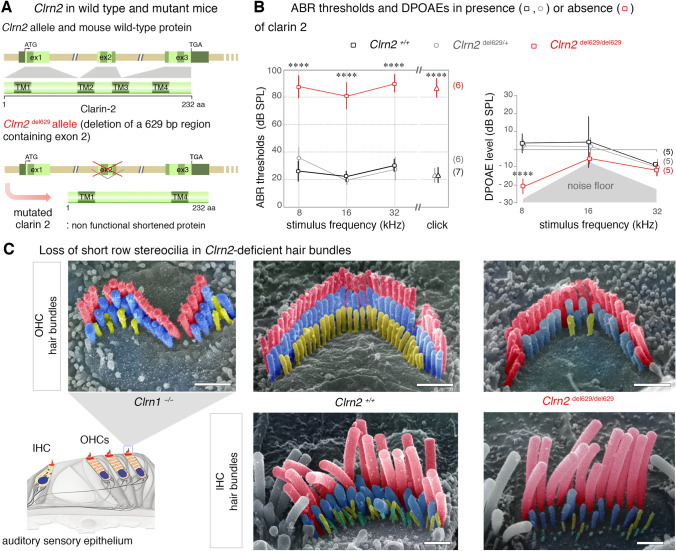
Deafness, the most frequent sensory deficit in humans, is extremely heterogeneous with hundreds of genes involved. Clinical and genetic analyses of an extended consanguineous family with pre-lingual, moderate-to-profound autosomal recessive sensorineural hearing loss, allowed us to identify CLRN2, encoding a tetraspan protein, as a new deafness gene. Homozygosity mapping followed by exome sequencing identified a 14.96 Mb locus on chromosome 4p15.32p15.1 containing a likely pathogenic missense variant in CLRN2 (c.494C > A, NM_001079827.2) segregating with the disease. Using in vitro RNA splicing analysis, we show that the CLRN2 c.494C > A variant leads to two events: (1) the substitution of a highly conserved threonine (uncharged amino acid) to lysine (charged amino acid) at position 165, p.(Thr165Lys), and (2) aberrant splicing, with the retention of intron 2 resulting in a stop codon after 26 additional amino acids, p.(Gly146Lysfs*26). Expression studies and phenotyping of newly produced zebrafish and mouse models deficient for clarin 2 further confirm that clarin 2, expressed in the inner ear hair cells, is essential for normal organization and maintenance of the auditory hair bundles, and for hearing function. Together, our findings identify CLRN2 as a new deafness gene, which will impact future diagnosis and treatment for deaf patients.
耳聋是人类最常见的感觉缺陷,其具有极高的异质性,涉及数百个基因。对一个具有语前、中度至重度常染色体隐性感觉神经性听力损失的近亲家族进行临床和遗传分析,使我们能够识别 CLRN2,其编码一种四跨膜蛋白,是一种新的耳聋基因。通过连锁分析和外显子组测序,我们确定了 4p15.32p15.1 染色体上的一个 14.96 Mb 的区域包含 CLRN2 中的一个可能致病的错义变异(c.494C > A,NM_001079827.2),该变异与疾病共分离。使用体外 RNA 剪接分析,我们表明 CLRN2 c.494C > A 变异导致两个事件:(1)在位置 165 处,将高度保守的苏氨酸(不带电荷的氨基酸)替换为赖氨酸(带电荷的氨基酸),p.(Thr165Lys);(2)异常剪接,内含子 2 保留,导致 26 个额外氨基酸后出现终止密码子,p.(Gly146Lysfs*26)。clarín 2 在表达研究和新产生的斑马鱼和小鼠模型中的表型缺失进一步证实,clarín 2 在内耳毛细胞中表达,对于正常的听觉毛束的组织和维持以及听力功能是必需的。总之,我们的发现确定了 CLRN2 是一种新的耳聋基因,这将影响未来耳聋患者的诊断和治疗。