Département D'immunologie et Biologie Cellulaire, Pavillon de Recherche Appliquée Sur le Cancer, Faculté de Médecine et Des Sciences de la Santé, Université de Sherbrooke, Sherbrooke, QC J1E 4K8, Canada.
Cells. 2021 Jan 23;10(2):224. doi: 10.3390/cells10020224.
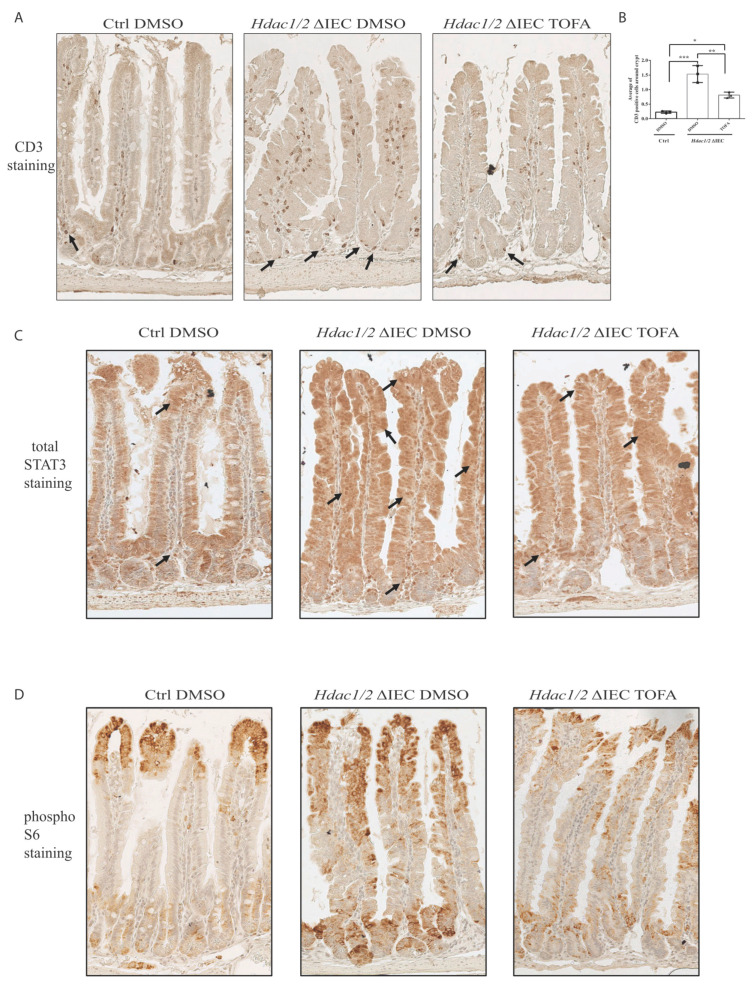
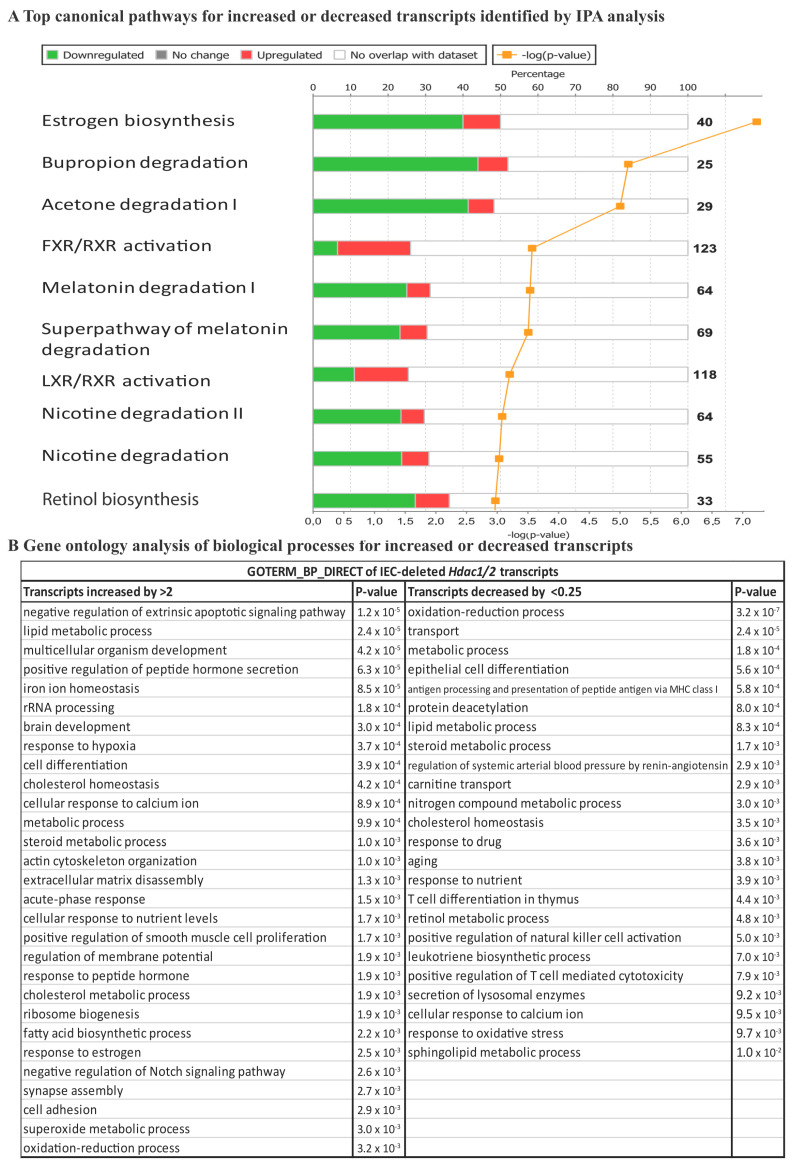
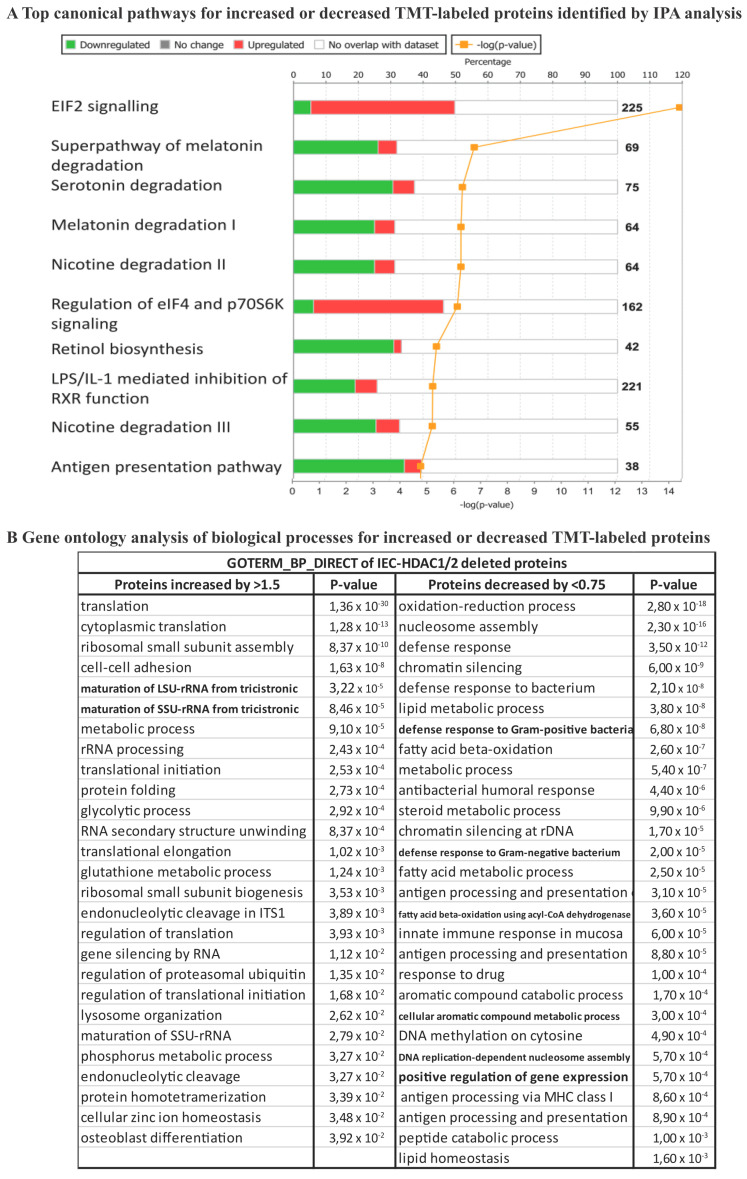
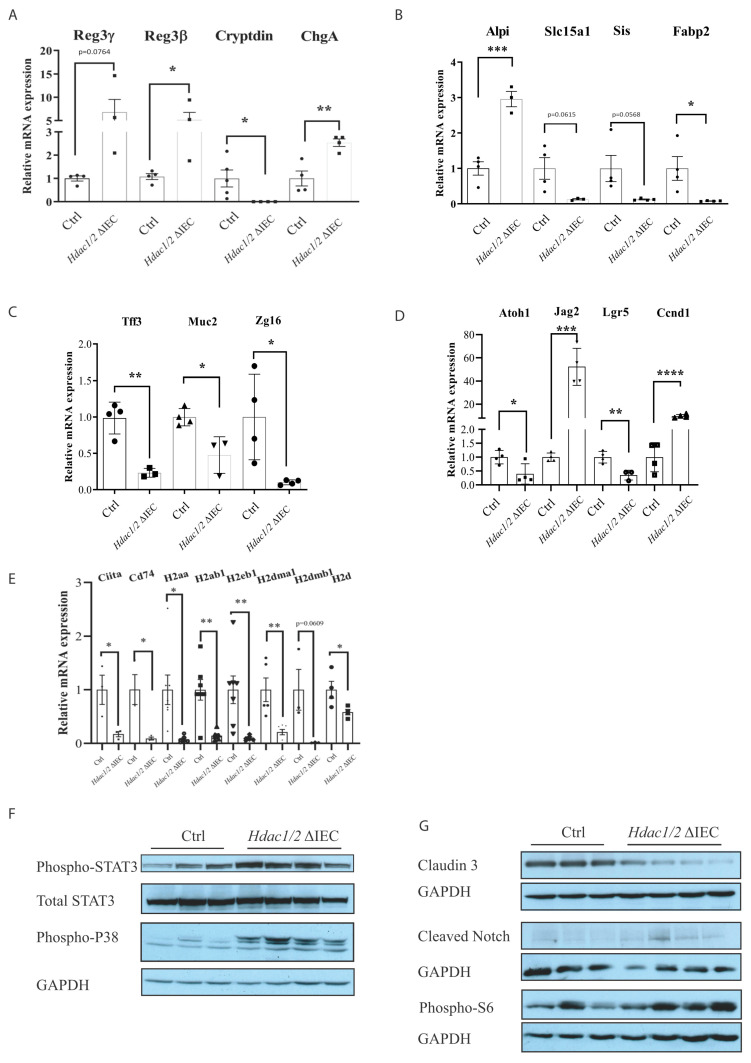
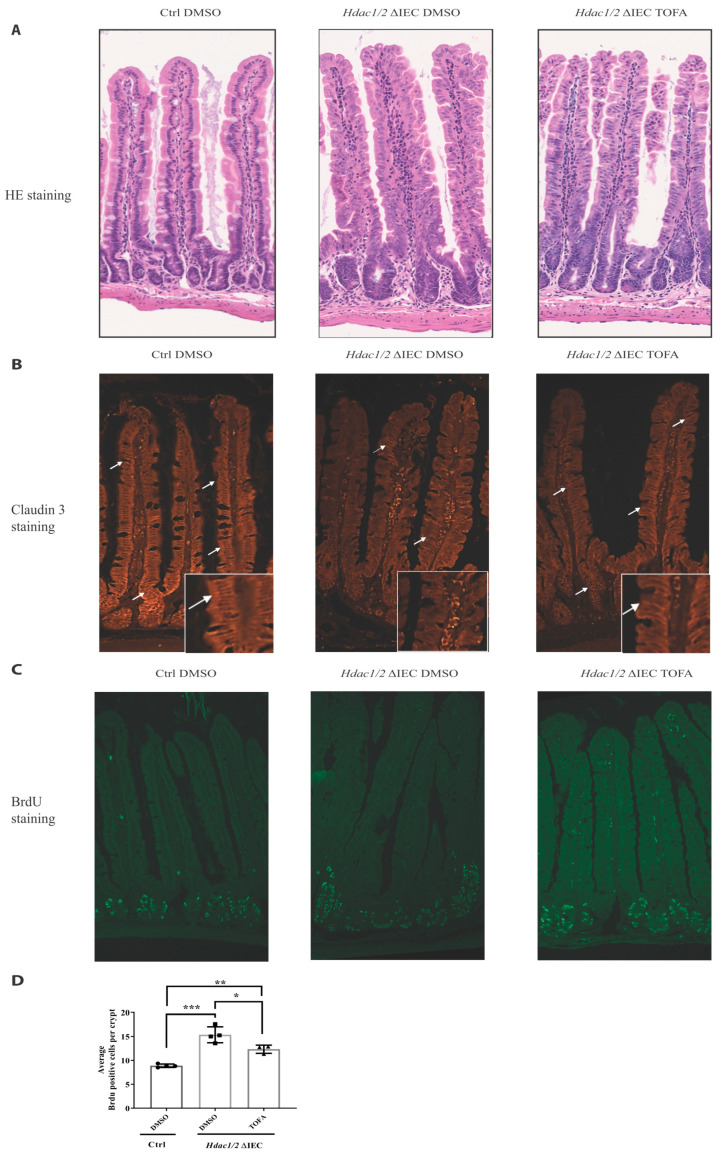
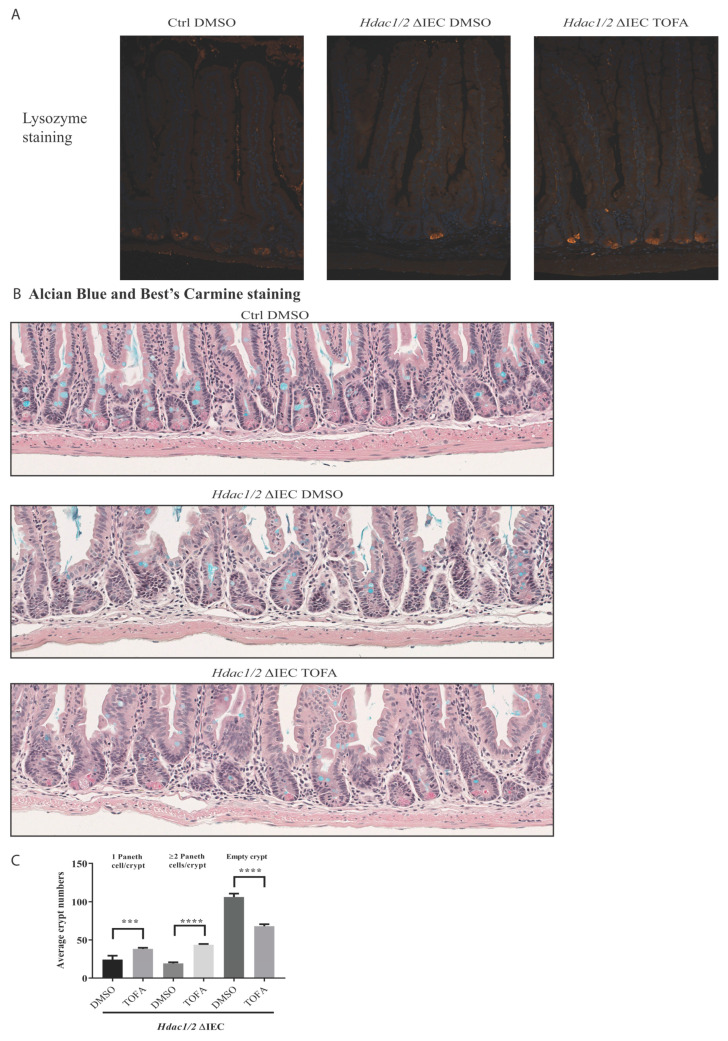
We have previously reported that histone deacetylase epigenetic regulator and deletion in intestinal epithelial cells (IEC) disrupts mucosal tissue architecture and barrier, causing chronic inflammation. In this study, proteome and transcriptome analysis revealed the importance of signaling pathways induced upon genetic IEC- and deletion. Indeed, Gene Ontology biological process analysis of enriched deficient IEC RNA and proteins identified common pathways, including lipid metabolic and oxidation-reduction process, cell adhesion, and antigen processing and presentation, related to immune responses, correlating with dysregulation of major histocompatibility complex (MHC) class II genes. Top upstream regulators included regulators associated with environmental sensing pathways to xenobiotics, microbial and diet-derived ligands, and endogenous metabolites. Proteome analysis revealed mTOR signaling IEC-specific defects. In addition to mTOR, the STAT and Notch pathways were dysregulated specifically in jejunal IEC. To determine the impact of pathway dysregulation on mutant jejunum alterations, we treated mutant mice with Tofacitinib, a JAK inhibitor. Treatment with the inhibitor partially corrected proliferation and tight junction defects, as well as niche stabilization by increasing Paneth cell numbers. Thus, IEC-specific histone deacetylases 1 (HDAC1) and 2 (HDAC2) support intestinal homeostasis by regulating survival and translation processes, as well as differentiation and metabolic pathways. HDAC1 and HDAC2 may play an important role in the regulation of IEC-specific inflammatory responses by controlling, directly or indirectly, the JAK/STAT pathway. IEC-specific JAK/STAT pathway deregulation may be, at least in part, responsible for intestinal homeostasis disruption in mutant mice.
我们之前曾报道过,组蛋白去乙酰化酶表观遗传调节剂和在肠上皮细胞(IEC)中的缺失破坏了黏膜组织的结构和屏障,导致慢性炎症。在这项研究中,蛋白质组和转录组分析揭示了遗传缺失诱导的信号通路的重要性。事实上,对基因缺失的 IEC RNA 和蛋白质进行的基因本体生物过程分析确定了共同的途径,包括与免疫反应相关的脂质代谢和氧化还原过程、细胞黏附和抗原加工和呈递,这与主要组织相容性复合体(MHC)Ⅱ类基因的失调有关。富集的缺陷 IEC RNA 和蛋白质的基因本体生物过程分析确定了共同的途径,包括与免疫反应相关的脂质代谢和氧化还原过程、细胞黏附和抗原加工和呈递,这与主要组织相容性复合体(MHC)Ⅱ类基因的失调有关。富集的缺陷 IEC RNA 和蛋白质的基因本体生物过程分析确定了共同的途径,包括与免疫反应相关的脂质代谢和氧化还原过程、细胞黏附和抗原加工和呈递,这与主要组织相容性复合体(MHC)Ⅱ类基因的失调有关。最重要的上游调节因子包括与环境感应途径相关的调节因子,这些途径与外源性化合物、微生物和饮食衍生配体以及内源性代谢物有关。蛋白质组分析显示 mTOR 信号在 IEC 中存在特异性缺陷。除了 mTOR 之外,STAT 和 Notch 途径在空肠 IEC 中也存在失调。为了确定通路失调对突变空肠改变的影响,我们用 JAK 抑制剂托法替尼(Tofacitinib)治疗突变小鼠。抑制剂的治疗部分纠正了增殖和紧密连接缺陷,并通过增加潘氏细胞数量稳定了小生境。因此,IEC 特异性组蛋白去乙酰化酶 1(HDAC1)和 2(HDAC2)通过调节存活和翻译过程以及分化和代谢途径来支持肠道内稳态。HDAC1 和 HDAC2 可能通过直接或间接控制 JAK/STAT 途径,在调节 IEC 特异性炎症反应中发挥重要作用。IEC 特异性 JAK/STAT 途径失调可能至少部分导致突变小鼠的肠道内稳态破坏。